1. Flask culture cell growth
- From frozen stocks, streak Salmonella serovar Typhimurium strains on LB agar (20 mL solid media in 100 mm Petri plate) with the appropriate antibiotic and incubate at 37 °C for 16–20 h to obtain isolated colonies.
- Inoculate 5 mL LB broth containing the appropriate antibiotic in a 25 mm x 150 mm borosilicate culture tube with 1–3 colonies from streak plates from step 1.1. Incubate at 37 °C with shaking at 200 rpm for 7 h.
- Measure OD600 of broth culture using a spectrophotometer. Dilute an aliquot of culture 1 in 4 in PBS in a 2 mL cuvette and measure the absorbance at 600 nm. We have found the most critical factor at this step to be the time of growth. The OD values are used to normalize the cultures to deliver the desired number of cells in step 1.4.
- Add 1.0 OD600 volume equivalent of cell culture (i.e. ~109 cells) to an Erlenmeyer flask containing 100 mL of 1% tryptone with the appropriate antibiotic. Incubate at 28 °C with shaking at 200 rpm for 13 h.
Note: Biofilm cells will appear as aggregated flakes and single planktonic cells are in the cloudy media.
2. Collecting cell-free conditioned 1% tryptone
- Collect flask culture for cell-free conditioned 1% tryptone. Pipette flask culture several times to mix before adding to a centrifuge tube.
- Centrifuge at 12,000 x g for 10 min at 10 °C to pellet all cells.
- Decant supernatant into a 0.2 µm filter unit, vacuum filter, and dispense into a new tube.
Note: Biofilm aggregates are adherent in conventional solutions (e.g. PBS) and will stick to the sides of tubes and pipette tips. Since it allows for easier manipulation, we use conditioned media (1% tryptone) to move biofilm initially and use warm PBS immediately before cross-linking (step 4.5) to remove any protein crosslinking substrates that could be present in the media.
3. Separating biofilm and planktonic cells from flask cultures2
- Using a sterile 25 mL pipette, aliquot flask culture into 15 mL tubes. Centrifuge at slow speed (210 x g) for 2 min, to separate the two cell types.
Note: Biofilm cells should form a loose pellet at the bottom of the tube - Pipette the supernatant containing planktonic cells into two 40 mL centrifuge tubes for later (step 5). Remove all the remaining liquid, being careful not to disturb the pellet. Repeat until the cell types have been separated from all of the flask culture media.
4. Preparing biofilm aggregates
Note: Biofilm is harvested by wet weight to yield 25 µg of DNA, the recommended minimum starting material for ChIP. The Biofilm Conversion Factor (BCF) of S. Typhimurium is 1.73 x 108 CFU/mg2. Researchers preparing other biofilm types will need to empirically define the number of cells per unit weight of biofilm, which is used to find the weight of biofilm required for 25 µg of DNA starting material using this equation:
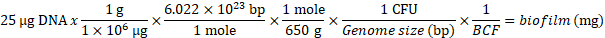
- Weigh a 2 mL snap cap or screw cap tube accurately.
- Resuspend the biofilm pellet in 1 mL of conditioned tryptone. Move the resuspended biofilm into a pre-weighed 2 mL snap cap or screw cap tube.
- Centrifuge for 1 min at 11 000 x g at 20 °C
- Carefully remove all supernatant from the tube and weigh the tube accurately. Subtract the tube weight from the weight of the tube with biofilm to find the weight of biofilm. It should be ±10% of the target biofilm weight.
- Wash cells to remove remaining conditioned tryptone. Add 1 mL of warm PBS to the tube and vortex gently to resuspend the pellet. Centrifuge for 3 min at 8 000 x g to pellet biofilm and remove the supernatant. Add 1 mL of PBS to the tube and vortex to resuspend the pellet.
5. Preparing planktonic cells
- Record the volume and the OD600 of the planktonic supernatant from slow speed centrifugation (step 3.2) using a spectrophotometer
- Calculate the required volume for a final OD600 of 6.0:
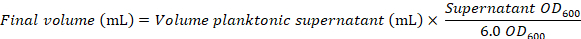
- Cool the centrifuge to 10 °C and sediment planktonic cells by centrifugation (10,000 x g, 10 min at 10 °C)
- Remove the supernatant and resuspend the cell pellet in the final volume of PBS calculated in step 5.2.
- Re-measure OD600 of the planktonic cells using a spectrophotometer and dispense the volume of 6.0 OD600 planktonic cells into a 2 mL snap cap or screw cap tube.
- Bring volume to 1 mL with PBS.
6. Homogenizing cells
- Add one sterilized 5 mm stainless steel bead to each of the tubes.
- Homogenize using a mixer mill for 5 min at 30 Hz. Observe aggregate tubes to confirm that biofilm has been broken apart.
- Transfer the homogenized cells to a new 1.5 mL tube, avoiding the metal bead. Bring the volume to 1 mL with PBS.
Note: Perform serial dilutions to enumerate input cells and check that the final cell number is close to the desired or predicted number.
7. Cross-linking of proteins to DNA
- Dispense fresh formaldehyde into sample tubes to a final concentration of 1%. Incubate for 30 min at room temperature on a rotating wheel.
Caution: Formaldehyde is corrosive, a skin, eye, and respiratory irritant, and flammable. Dispense in a fume hood. - Add 1 M glycine to a final concentration of 125 mM to stop crosslinking. Incubate for 5 min at room temperature on a rotating wheel.
8. Washing cells to remove excess crosslinker
- Sediment the cells at 8,000 x g for 3 min and remove the supernatant
- Resuspend the pellet in 20 µL of 25x protease inhibitors and 500 µL of filter sterilized PBS.
- Centrifuge tube for 3 min at 8000 x g and remove the supernatant.
9. Lysing cells
- Resuspend the pellet in 600 µL Lysis buffer (refer to Table 1) and incubate on ice for 10 min.
- Add 1.4 mL of IP dilution buffer (refer to Table 1) to a sterile 15 mL tube and move lysed cells to the 15 mL tube. Keep the tube on ice for 1.5–2 h, and vortex gently and occasionally.
Note: Some resistant cell material may remain in tubes. A long incubation period on ice with occasional vortexing will break apart material. The remainder will be broken up through sonication.
10. Fragmenting DNA by sonication
- Place the 15 mL tube in a beaker of ice and place the 3 mm probe inside the tube.
- Perform 5 sonication rounds of 30 s on at 20–40% of 400 W with 2 min cooling on ice between bursts.
- Sediment precipitated material by centrifugation (8000 x g, 10 min at 4 °C). Transfer the supernatant to a new tube.
- Optionally, perform decrosslinking at 65 °C for 30–60 min and digest RNA and protein at 45 °C for 30-60 min before running on a 2% agarose gel to check for proper size fragments.
Note: Immerse the probe into the cell lysate. Keep the solution on ice while sonicating and resting. Do not remove the tube while the sonicator probe is pulsing. The solution should appear cloudy pre-sonication and clear post-sonication.
11. Immunoprecipitating DNA-protein-antibody complexes
- Preparation
- Dispense one 1.35 mL aliquot for immunoprecipitation and one 200 µL aliquot as an input control. Store the input control at -80 °C until decrosslinking and digesting steps are performed.
- Immunoprecipitation with primary antibody
- Add 10 µg of purified protein-specific primary antibody to the 1.35 µL aliquot. Incubate overnight at 4 °C on a rotating wheel.
Note: The primary antibody can be a monoclonal antibody, high-quality polyclonal serum or commercial epitope antibody (i.e., anti-FLAG). - Add 50 µL of Protein G magnetic beads to the tubes from step 11.2.1 and incubate at 4 °C for 3 h on a rotating wheel.
- Place IP wash buffer 1, IP wash buffer 2, and TE pH 8.0 (refer to Table 1) in an ice bucket. Warm elution buffer to 65 °C.
Note: Do not put solution containing elution buffer on ice. - Bind the Protein G magnetic beads to the side of tubes using a magnetic stand
- Perform washes: wash 2x with 750 µL cold IP wash buffer 1, wash 1x with 750 µL cold IP wash buffer 2, and wash 2x with 750 µL cold TE at pH 8.0. Keep the tubes on the magnetic stand during washes.
- Add 450 µL of IP Elution buffer (refer to Table 1) to each tube and incubate at 65 °C for 30 min with gentle vortexing every 5 min.
- Bind beads to the side of tubes using a magnetic stand. Wait at least 2 min until the solution is clear.
- Dispense the cleared supernatant to a new 1.5 mL tube, being careful to not disturb the magnetic bead pellet.
- Add 10 µg of purified protein-specific primary antibody to the 1.35 µL aliquot. Incubate overnight at 4 °C on a rotating wheel.
12. Decrosslinking DNA-protein complexes and digesting co-purifying RNA and protein
- Add 2 µg RNase A and NaCl to a final concentration of 0.3 M to each tube.
- Incubate at 65 °C, for ≥6 h, or overnight.
- Complete protein digestion by adding 180 µg Proteinase K to each tube, then incubating at 45 °C for 3–5 h.
13. Library preparation and sequencing
- Purify DNA using magnetic beads
Note: Magnetic beads are preferred for isolating the small amounts of DNA usually recovered during ChIP with bacterial cells. - Prepare libraries using a kit that is compatible with the selected sequencing platform.
- Check library concentration with a fluorometer and a broad range dsDNA kit or a qRT-PCR library quantification kit.
Note: In our experience, fluorometer measurements may overestimate DNA concentration. - Pool libraries, accounting for adequate coverage for each library sample. For Illumina sequencing with Salmonella, we pooled 10–12 libraries. Add Tris-HCl pH 8.0 to a final concentration of 1 mM.
- Sequence according to the selected platform's specifications.
Preparation of chromatin immunoprecipitation samples from bacterial biofilm involves several steps, as shown in Figure 1. These include growing biofilm in flask culture, collecting conditioned media, collecting an adequate amount of biofilm and planktonic cells as control, washing cells in PBS, homogenization, crosslinking proteins to DNA, lysing cells, fragmenting DNA by sonication, immunoprecipitating DNA with the appropriate antibodies and protein G beads, washing and eluting immunoprecipitated DNA, and purifying DNA for library preparation and sequencing.
S. Typhimurium cells grown in liquid culture under biofilm-inducing conditions undergo phenotype switching to form multicellular biofilm aggregates and planktonic cells. This population will contain approximately 40% biofilm aggregates, which express higher levels of diguanylate cyclases (DGCs) and biofilm regulators, and 60% planktonic cells, which express higher levels of virulence-associated genes2,4 (Figure 2A). In this protocol, we describe a method to harvest both cell types to compare transcription sites through ChIP-seq; however, this protocol can be adapted for other types of biofilms formed by other bacterial species. Biofilm aggregates and planktonic cells are separated by slow speed centrifugation, which pellets biofilm and leaves planktonic cells in the supernatant2 (Figure 2B). The cell types are then treated separately for subsequent steps in the protocol.
The recommended amount of DNA input for ChIP-seq is 10-25 µg20. In S. Typhimurium flask culture, 30 mg of biofilm yields approximately 25 µg DNA. Since biofilm aggregates have an abundance of proteinaceous extracellular material, we hypothesized that this would interfere with the efficiency of cross-linking. If we were simply to normalize the different cell types by cell number, treatment of planktonic cells may result in an unequal amount of crosslinked product presented for immunoprecipitation. A protein assay was performed to determine the equivalent amount of planktonic cell material to match 30 mg of biofilm (Figure 3). 6.0 OD600 of planktonic cells were harvested to match 30 mg biofilm.
Biofilm aggregates must first be broken apart to allow equal access of cross-linker to all cells. We found that the most uniform and high-throughput way to homogenize the cells was using a mixer mill and 5 mm stainless steel bead (Figure 4A). Planktonic cells are treated the same way to reduce variables in the ChIP-seq experiment (Figure 4B).
Once the DNA has been fragmented by sonication, crosslinked, and treated with RNase and Proteinase K, it can be visualized on a 2% agarose gel. Sonicated DNA is broken down into a collection of fragments that appear as a smear on the agarose gel. The average fragment size decreases with an increasing number of sonication bursts (Figure 5). Energy transfer will vary for different sonicator units, so a sonication assay is recommended to determine the number of sonication bursts required to fragment DNA to an average of less than 1 kb. For our ChIP-seq experiment, we chose 5 bursts.
ENCODE guidelines recommend a confirmation of antibody target-binding activity with an immunoblot21. In this case we measured our antibody's specificity and strength of binding by immunoblot on whole cell lysates prepared for ChIP (Figure 6). We confirmed that the antibody binds to CsgD-3xFLAG; anti-FLAG and anti-CsgD signals colocalize at the expected molecular weight (28 kDa)22.
ChIP procedures often yield low concentrations of DNA. Therefore, a purification method that removes contaminants and retains a high proportion of the DNA was chosen. Magnetic bead purification was chosen over column-based purification because it retained low concentrations of input ChIP DNA better (Figure 7) and removed contaminants (data not shown).
Due to reduced starting amounts of DNA, library preparation of ChIP DNA may require diluted adapters and PCR enrichment. Prior to sequencing, libraries can be visualized by Bioanalyzer trace to confirm correct size distribution before and after PCR enrichment (Figure 8). Additionally, if there is a known regulatory target of the transcription factor, qPCR should be used to confirm enrichment of binding regions by comparing fold change (∆∆Ct) between known target and reference gene sequence abundance in immunoprecipitated and sonicated input DNA.
ChIP-sequencing data should be analyzed by assigning quality scores, trimming low quality bases, aligning forward and reverse reads (in paired end sequencing), assembling to a high-quality reference genome, finding peaks above background, and performing downstream analysis (Figure 9A). Downstream analysis should include visualization and annotation of peaks, consistency with biological replicate samples, and motif analysis, and could include differential binding analysis between conditions or cell types, and data integration with gene expression from known pathways. For ChIP-seq data such as ours, peaks should be identified as significantly above background (Figure 9B,C, red bars) with a p value determination, not simply by fold-change. In the final analysis, the mapped reads are visualized against the annotation of the reference genome (Figure 9D). A poor ChIP-seq result would appear like input-seq without any significant peaks above background.
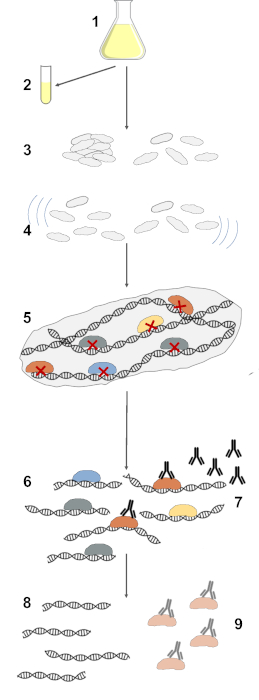
Figure 1: Illustration of steps in ChIP-seq with bacterial biofilms. In the described procedure, S. Typhimurium cells are grown in 1% Tryptone flask culture at 28 °C with shaking (1), cell-free conditioned media is collected for handling biofilm cell types (2), which is used to collect an adequate amount of biofilm and planktonic cells (3). These cells are washed with PBS and homogenized to break apart biofilm (4). Proteins are crosslinked to DNA with a formaldehyde crosslinker, after which cells are lysed to release cell contents (5). DNA in the cell lysate is fragmented by sonication (6). DNA crosslinked to the target protein is selected through immunoprecipitation with an antibody that binds to the target protein and Protein G magnetic beads (7). Selected DNA is washed and eluted from Protein G magnetic beads (8) and purified for library preparation and sequencing (9). Please click here to view a larger version of this figure.
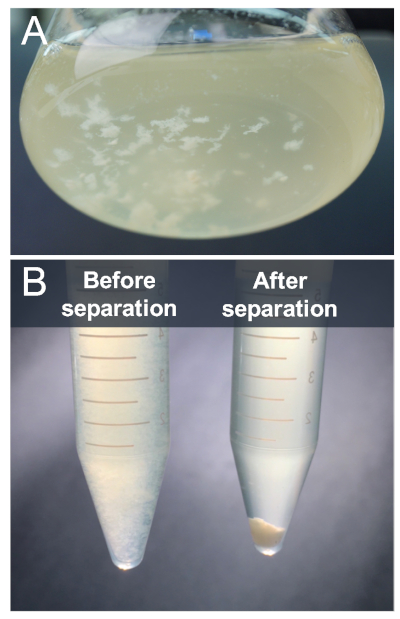
Figure 2: In vitro flask model for studying S. Typhimurium biofilm development. (A) S. Typhimurium cells grown in 1% tryptone for 13 h at 28 °C undergo phenotype switching to form biofilm aggregates and planktonic cells in the liquid phase of the flask culture. (B) Biofilm aggregates and planktonic cells from the flask culture in (A) were separated through centrifugation at 210 x g for 2 min. The supernatant was harvested for planktonic cell preparations and the pellet was harvested for biofilm cell preparations. Please click here to view a larger version of this figure.
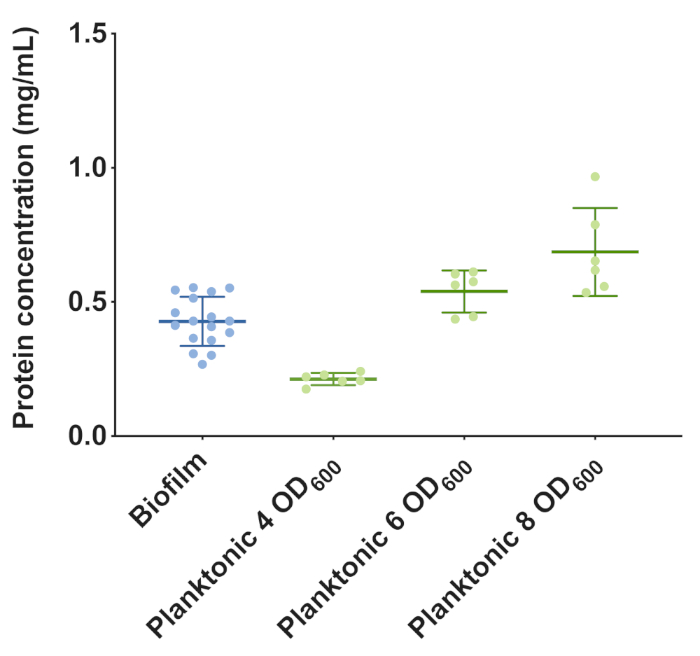
Figure 3: Total protein concentrations for planktonic cells and biofilm cell samples harvested from S. Typhimurium flask culture. Colorimetric protein assays were performed and protein amounts relative to a BSA standard curve were measured at 750 nm. Protein content of lysed planktonic cell samples at 4.0, 6.0, and 8.0 OD600 were compared to 30 mg of biofilm aggregates. Please click here to view a larger version of this figure.

Figure 4: Homogenization of cell samples from S. Typhimurium biofilm flask cultures. (A) Biofilm aggregates from flask culture resuspended in PBS (left) were homogenized using a mixer mill at 30 Hz for 5 min (right). (B) Planktonic cells from flask culture resuspended in PBS were processed by mixer mill at 30 Hz for 5 min to ensure consistency between cell type samples. Please click here to view a larger version of this figure.
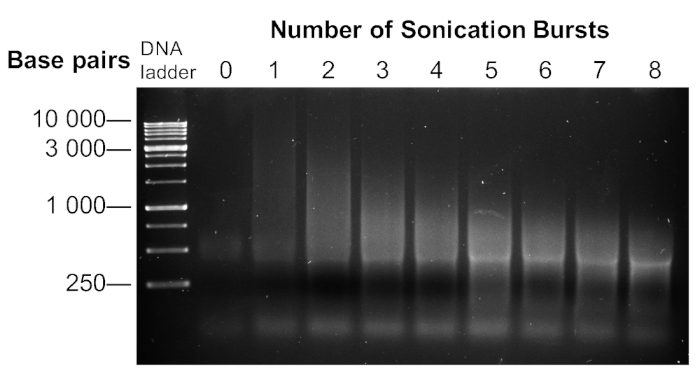
Figure 5: Sonication assay with pre-ChIP cell lysates. Biofilm aggregate and planktonic cell samples were separated, homogenized, crosslinked, and lysed. DNA-protein complexes were sonicated up to 8 times at 30 s on and 2 min off on ice to break DNA into smaller fragments. A portion of the sonicated lysate was separated on a 2% agarose gel. Please click here to view a larger version of this figure.
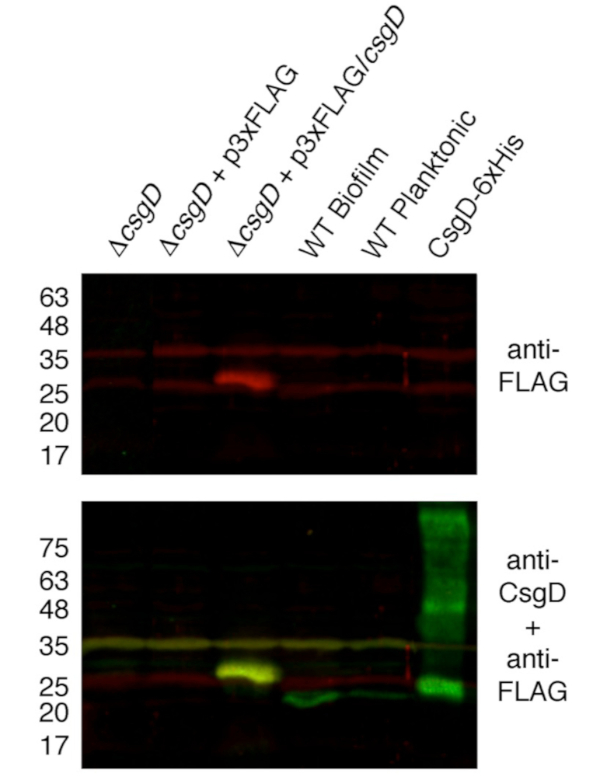
Figure 6: Target-binding specificity of anti-FLAG antibody used in ChIP-seq. Antibody specificity was assessed by immunoblotting against whole cell lysates prepared from flask cultures of the S. Typhimurium strains as shown. Strain ∆csgD + p3xFLAG/csgD and WT biofilm samples represent biofilm aggregates harvested from flasks, whereas strain ∆csgD ± p3xFLAG and WT planktonic cells represent planktonic cells. Purified His-tagged CsgD was loaded as a control. Whole cell lysates were normalized so that 30 µg of total protein was analyzed in each lane. Numbers on the left refer to the size (in kDa) of the prestained molecular weight markers. Primary antibody used for the upper blot was rabbit-anti-FLAG polyclonal antibody (Sigma-Aldrich #F7425), whereas the lower blot was incubated with rabbit-anti-FLAG together with a CsgD-specific monoclonal antibody2. Goat anti-rabbit IgG (LiCor IRDye 680RD, 925-68071) and goat anti-mouse IgG (LiCor IRDye 800CW, 925-32210) were used as secondary antibodies. Fluorescent signals were visualized using the Odyssey CLx imaging system and Image Studio 4.0 software package (Li-Cor Biosciences). Representative images are shown. Please click here to view a larger version of this figure.
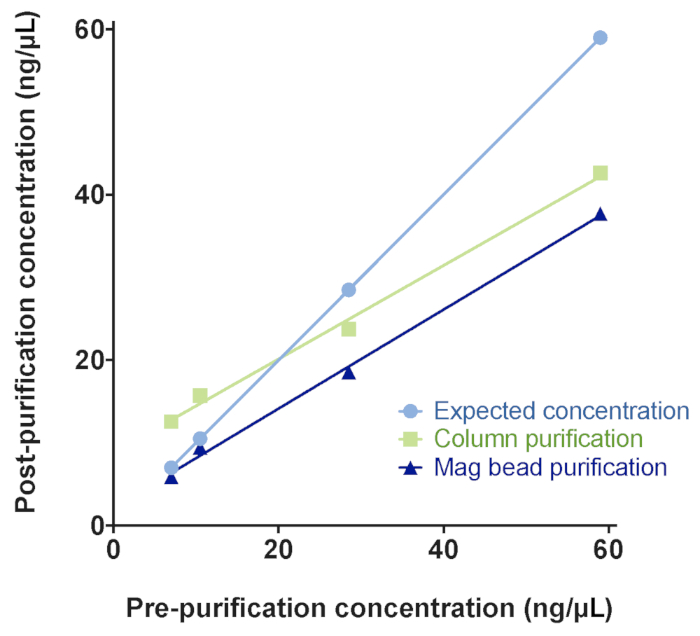
Figure 7: Column- or magnetic bead-based purification strategies for small amounts of DNA resulting from ChIP-seq experiments. Samples of known quantities of DNA were purified using column-based kits or magnetic beads and the concentration of the eluate was measured post-purification. Magnetic beads showed better recovery at low DNA levels, similar to the low amounts of DNA typically encountered at the end of ChIP-seq protocol, but prior to NGS library preparation. Please click here to view a larger version of this figure.
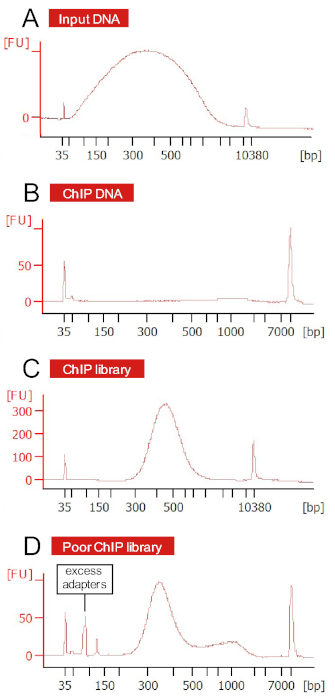
Figure 8: ChIP DNA preparations and subsequent libraries visualized by Bioanalyzer. (A) Average fragment size of genomic DNA after cell lysis and sonication was <1000 bp, with a majority of fragments in the 200–500 bp range. (B) Starting material for library preparation was below detection. (C) Adapter ligation and PCR enrichment allows for amplification of library fragments. (D) A poor library preparation has low DNA peaks, odd shaped or high molecular weight peaks, or abundant adapter peaks at approximately 100 bp. Please click here to view a larger version of this figure.
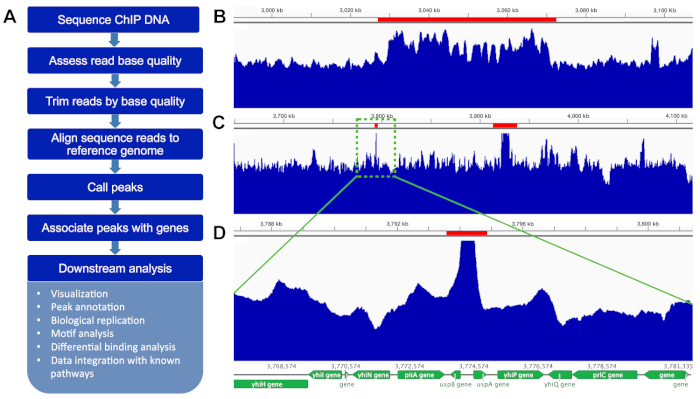
Figure 9: ChIP-seq data is analyzed using bioinformatic tools and visualized on a genome browser. A. Flowchart for typical bioinformatic analysis of ChIP-seq data. Raw reads for biofilm (∆csgD strain + p3xFLAG/csgD) and planktonic cells (∆csgD + p3xFLAG) were cleaned for low-quality reads and adapters, and mapped to S. Typhimurium 14028s genomes (NC_016856.1 for the chromosome and NC_016855.1 for the plasmid) by Bowtie2 (v2.3.3.1) with default parameters23. MACS2 (v2.1.2) was used to call the peaks with parameters of '-q 0.01 — nomodel', taking biofilm replicates as testing datasets and planktonic ones as control24. The MACS2 bdgcmp module was further used to generate fold-enrichment track with parameters of '-m FE'. The significant peak file with fold-enrichment track was transformed to a wig file using bedtools/bedClip/bedGraphToBigWig25 and visualized on the reference genome using Integrative Genomics Viewer v2.5.126. Representative peaks are shown (red bars). (B) Large region of ~40 kbp, which contains multiple peaks, is an atypical result but still potentially valuable. (C) This is a more typical result, showing two significant peak regions. (D) Zoom in on left peak in C, showing reads mapping to the region of the S. Typhimurium 14028s genome containing divergent promoters for uspA and uspB. Please click here to view a larger version of this figure.
| Cell lysis buffers |
| Lysis buffer |
| 50 mM Tris-HCl pH 8.1 |
| 10 mM EDTA |
| 1% SDS |
| protease inhibitors |
| IP dilution buffer |
| 20 mM Tris-HCl pH 8.1 |
| 2 mM EDTA |
| 150 mM NaCl |
| 1% Triton X-100 |
| 0.01% SDS |
| Immunoprecipitation buffers |
| IP wash buffer 1 |
| 20 mM Tris-HCl pH 8.1 |
| 2 mM EDTA |
| 50 mM NaCl |
| 1% Triton X-100 |
| 0.1% SDS |
| IP wash buffer 2 |
| 10 mM Tris-HCl pH 8.1 |
| 250 mM LiCl |
| 1 mM EDTA |
| 1% NP-40 |
| 1% deoxycholic acid |
| TE (T10E1) pH 8.0 |
| 10 mM Tris-HCl |
| 1 mM EDTA |
| Elution buffer |
| 1% SDS |
| 100 mM NaHCO3 |
Table 1. Buffer recipes.
