In Figure 2A, the typical pyramidal neurons in the hippocampal region in the rat brain sections were identified by ballistic labeling technology, characterized by one large apical dendrite and several smaller basal dendrites around the soma. Figure 2B shows the neuron in the neuronal reconstruction quantitative analysis software after the soma was detected, dendritic branches were traced, and spines were detected. Subsequently, the data were analyzed using neuronal reconstruction quantitative analysis software, which provided an opportunity to assess the dendritic branching complexity (Figure 2C) and neuronal arbor complexity (Figure 2D).
In Figure 2C, we utilized the centrifugal branch ordering method, collected from the “Tree Totals” output, to count the number of segments traversed along each dendrite and assigned branch order. The relative frequency of segments at each branch order was examined for branch orders 1–15. When shifts in the distribution of dendritic branches were observed between groups, alterations in dendritic branching complexity could be inferred. Furthermore, a Sholl analysis was conducted as a complementary measure of neuronal arbor complexity, whereby the number of dendritic intersections occurring every 10 µm from the soma was quantified in each sample section (Figure 2D). When shifts in the number of dendritic intersections were observed between groups, alterations in neuronal arbor complexity could be inferred.
Morphological changes in dendritic spines could be assessed using length (µm), head diameter (µm), and volume (µm3), as seen in Figure 3A–B. Furthermore, spines were classified using the automatic assisted classification system in the neuronal reconstruction software. The relative frequency of the number of spines between each radius was examined for thin, mushroom, and stubby spines. Given our understanding of which spine types form stronger synaptic connections (i.e., mushroom relative to stubby) and neurotransmitter afferents, shifts in the distribution of spines along the neuron can indicate synaptic connectivity.
Furthermore, we validated the utility of the ballistic labeling technique on a primary pyramidal neuron in cell culture. First, we cultured primary hippocampal neurons at postnatal D1 (day 1) on a cell culture plate coated with poly-L-lysine for 2 weeks or until approximately 70% confluency. Then the samples were fixed with 4% PFA for 15 min and washed 2x with PBS. Beginning at step 3.1 of the present protocol, we ballistically labeled and imaged the primary pyramidal neurons from the hippocampus. Data showed stabilized labeling and identified pyramidal neurons based on the triangle shape of the soma and large apical dendrite (Figure 4A). Utilization of neuronal reconstruction software to investigate dendritic spines of primary pyramidal neurons grown in cell culture offers opportunities similar to those in the rat brain. Example outcomes are illustrated for the distribution of thin dendritic spines (Figure 4B) and dendritic spine length measured in µm (Figure 4C). However, it is noteworthy that the primary pyramidal neurons grown in cell culture had less dendritic branching, precluding the assessment, at least in this example, of dendritic branching complexity and neuronal arbor complexity.
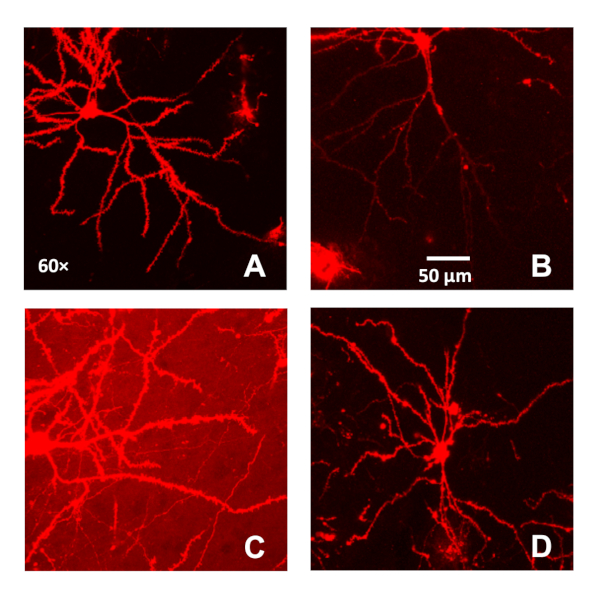
Figure 1: Selection criteria utilized for pyramidal neurons in the medial prefrontal cortex labeled using ballistic labeling technology. (A) A representative confocal image (60x) of a well-labeled pyramidal neuron from the medial prefrontal cortex. A single pyramidal neuron with a clear soma and apical dendrite included continuous, bright dendritic staining with low background. (B–C) A representative confocal image (60x) of a pyramidal neuron from the medial prefrontal cortex with light staining at the more distal branches (B) and high background (C). (D) A representative confocal image (60x) of a labeled neuron from the medial prefrontal cortex (based on Bregma coordinates) that has flawed morphological characteristics. Please click here to view a larger version of this figure.
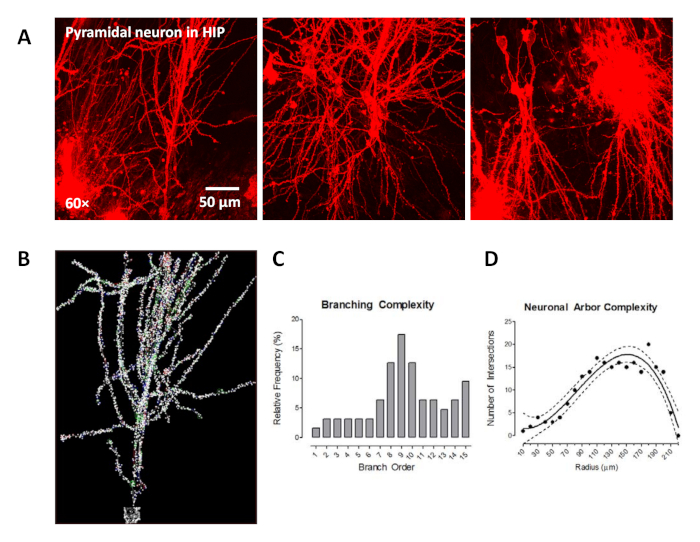
Figure 2: The labeling of pyramidal neurons in the hippocampus (HIP) using ballistic labeling technology and neuroanatomic assessments. (A) Three representative confocal images (60x) of pyramidal neurons labeled by ballistic tungsten beads. (B) The assessment of neuronal morphology: dendritic branch order analysis and Sholl analysis. The traced image of the dendritic spine in which the spine morphology was also identified using dendritic spine analysis software. (C) Branch order analyses utilized to examine the relative frequency of dendritic branches at different branch orders. (D) The numbers of dendritic intersections every 10 µm from the soma were assessed using Sholl analysis as a measure of neuronal arbor complexity. Data are described as relative frequencies of the entire dataset (C) or fit with 95% confidence intervals (D). Please click here to view a larger version of this figure.
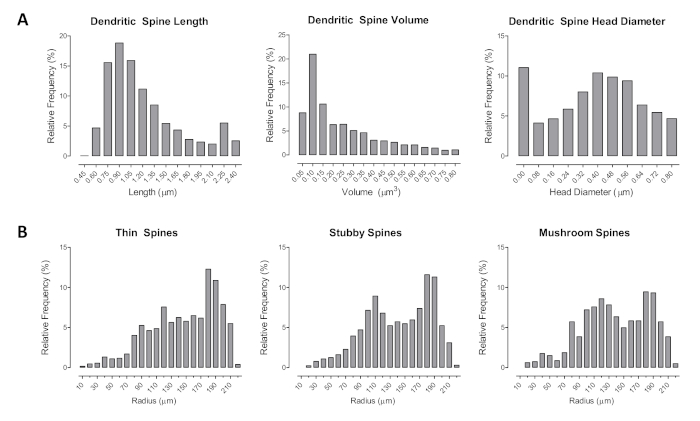
Figure 3. The assessment of dendritic spine morphology. (A–B) The distribution of dendritic spines illustrated as a function of spine type (i.e., thin, stubby, mushroom). Additional dendritic spine parameters were also analyzed (i.e., length, volume, head diameter) as an assessment of dendritic spine morphology. Data are illustrated as relative frequencies of the entire dataset. Please click here to view a larger version of this figure.
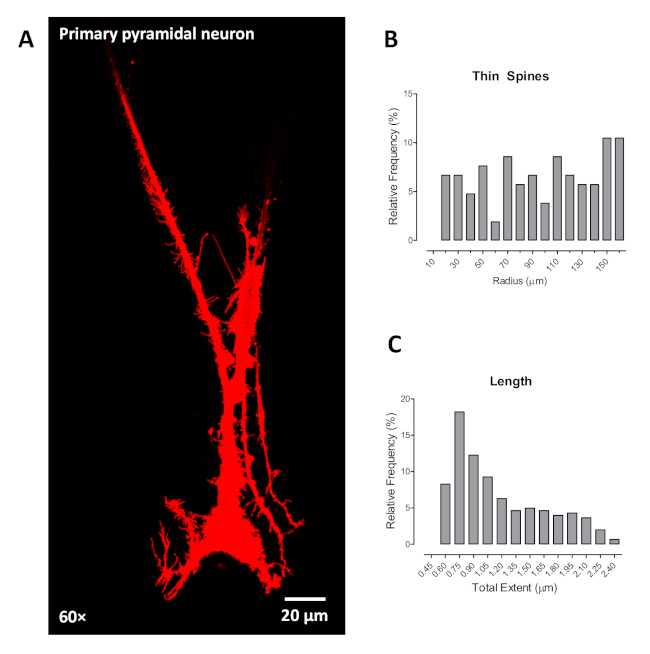
Figure 4. The labeling of primary cortical neuron in vitro using ballistic labeling technology. (A) Representative confocal images (60x) of primary cortical neurons labeled by ballistic tungsten beads in vitro. Example results similar to those acquired from ballistic labeling in brain slices are shown for the distribution of thin dendritic spines (B) and length (C). Data are illustrated as relative frequencies of the entire dataset. Please click here to view a larger version of this figure.
Video 1: Procedures of neuronal tracing and dendritic spine detection. Please click here to view this video. (Right-click to download.)
Video 2: Procedures of data collection and output for quantitative analysis. Please click here to view this video. (Right-click to download.)
