The work involving primary monocytes isolated from human blood was approved by the committee of the Federal Office for Public Health Switzerland (reference number: 611-1, Meldung A110635/2) for the Adolphe Merkle Institute.
1. Isolation of peripheral blood monocytes (PBMs) from human buffy coats
NOTE: The following section describes the isolation of immune cells from one 50 mL bag of a buffy coat, purchased from the Swiss Transfusion Centre in Bern, Switzerland.
- Preparation of reagents
- Prepare 100 mL of magnetic separation buffer per buffy coat: 0.5% [w/v] bovine serum albumin (BSA; in phosphate-buffered saline [PBS]) with 2 mM ethylenediaminetetraacetic acid (EDTA), and adjust to pH = 7.2, sterile filter with 0.22 µm pore size). Keep at 4 °C throughout the procedure.
- Prepare the cell culture medium (CCM): RPMI 1640 with 10% [v/v] fetal bovine serum (FBS), 1% [v/v] L-glutamine (here, 2 mM L-glutamine), and 1% [v/v] penicillin-streptomycin (here, 100 units/mL penicillin and 100 µg/mL streptomycin).
NOTE: The required amount of each reagent depends on the number of cells to be seeded in the following steps.
- Isolation of PBMs
NOTE: All glass and plasticware needs to be sterilized before use. For safety reasons, the use of plasticware is recommended when handling human blood samples to reduce the risk of injury with glassware.- Use scissors to cut open the hose end of the bag containing the buffy coat.
- Distribute the buffy coat by pouring the bag's contents through the bag's duct directly into two conical centrifuge 50 mL tubes (~25 mL each).
- Gently pour or pipette PBS into the tubes to reach 50 mL volumes. Mix the contents by turning the tube gently upside down 3x.
- Divide the buffy coat-PBS mixture into four new 50 mL conical centrifuge tubes by pipetting 25 mL of the mixture in each fresh tube.
- Slowly lay 13 mL of density gradient medium underneath the buffy coat-PBS mixture using a 10 mL serological pipette. Detach the filled pipette from the pipette holder and immediately plug the upper opening of the pipette with a thumb to prevent any additional leaking of the density gradient medium into the buffy coat-PBS mixture.
NOTE: Holding the upper opening with the thumb, place the filled pipette at the bottom of the conical centrifuge tube so that the density gradient medium slowly flows beneath the buffy coat-PBS mixture, leaving approximately 1 mL of the density gradient medium inside the pipette. - Repeat step 1.2.5 with the other three tubes containing the buffy coat-PBS mixture.
- Centrifuge all four tubes containing the mixtures for 20 min at 1,000 x g and 25 °C at a slow braking mode. Use holders with protective lids for centrifugation.
- Open the lid of each tube, remove the upper layer containing plasma and platelets using a serological pipette, and dispose in a biohazard liquid waste container.
- Use a serological pipette for collecting the peripheral blood mononuclear cell layer, which appears as a whitish turbid small fraction (~2–3 mm in thickness) between the plasma and the density gradient medium layers. The pellet contains red blood cells at the bottom. Avoid transferring erythrocytes that form the bottom-most layer. Repeat this for all four tubes.
NOTE: Peripheral blood mononuclear cells consist of PBMs and lymphocytes. PBMs will be separated from lymphocytes later during magnetic CD14+ separation. - Pool peripheral blood mononuclear cells from four tubes into two 50 mL tubes.
- Fill up the two tubes with PBS to 50 mL and cover with a lid.
- Discard the leftover erythrocytes and plasma from the original four tubes in a biohazard liquid waste container.
- Centrifuge the two tubes for 8 min at 500 x g and 18–20 °C at a regular centrifuge speed.
- After centrifugation, remove the supernatant with a serological pipette and discard it in a biohazard liquid waste container.
- Resuspend the cells with 5 mL of PBS using a serological pipette.
- Pool the cell suspensions into one 50 mL conical centrifuge tube and fill to 50 mL with PBS.
- Use 5 µL of the cell suspension to count the cells with a cell counter using the trypan blue (45 µL) exclusion method.
- Pipette 10 µL of the trypan blue-PBMs solution into a cell counter chamber and count the number of cells per the standard counting protocol. Use Equation 1 to calculate the total cell number, CT.
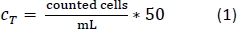
- After counting the cells, centrifuge the 50 mL tube as done in step 1.2.13.
- CD14 positive selection
- Gently open the lid of each tube, then remove and discard the supernatant using a serological pipette without disturbing the pellet.
- Add the calculated amount (Equation 2) of magnetic separation buffer (here, 80 µL of buffer per 1 x 107 total cells) and resuspend the cell pellet by pipetting the solution up and down.
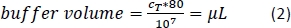
- Calculate using Equation 3 (here, 10 µL per 1 x 107 total cells) the corresponding volume of CD14+ magnetic beads and pipette the appropriate volume.
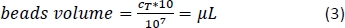
- Mix well by pipetting up and down, close the lid, and incubate the solution at 4 °C for 15 min.
- Upon incubation, fill the tube up to 50 mL with magnetic separation buffer.
- Centrifuge as done in step 1.2.13.
- Aspirate and discard the supernatant using a serological pipette without disturbing the cell pellet.
- Pipette the corresponding amount of magnetic separation buffer (Equation 4; here, 500 µL of buffer per 1 x 108 cells) and gently mix by pipetting up and down 3x.
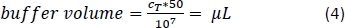
- Disinfect the magnetic separation station by spraying and wiping it with a sterilizing agent. Place in the laminar flow hood along with the column for magnetic separation.
- Place the magnetic separation column in the magnetic field and place an empty 50 mL conical centrifuge tube directly under the column for collecting the wash and unlabeled cells (i.e., waste).
- Rinse the magnetic separation column by pipetting 3 mL of magnetic separation buffer into the column. Do not let the column dry out throughout the procedure.
- Prepare a 15 mL conical centrifuge tube and pipette 1 mL of magnetic separation buffer.
- Apply the cell suspension (prepared in step 1.3.8) to the magnetic separation column. In the 50 mL conical centrifuge tube under the filter, collect unlabeled cells that passed through.
NOTE: Do not exceed 2 x 109 cells per column to avoid blocking of the column. - As soon as the column reservoir is empty (i.e., when the cells have passed through the column), apply 3 mL of magnetic separation buffer using a serological pipette and let it pass through the column. Repeat this 3x.
- Remove the magnetic separation column from the magnetic separator by gently pulling with hands, then place it into a 15 mL tube containing pre-pipetted 1 mL of magnetic separation buffer (prepared in step 1.3.12).
- Add 5 mL of magnetic separation buffer to the column and flush out the magnetically labeled cells by firmly pushing the plunger into the column.
- Reagent preparation for MDM and MDDC differentiation
- Count the cells with a cell counter using the trypan blue exclusion method as done in step 1.2.17.
- Calculate the required volumes of CCM or FBS for further steps as follows: either the volume of CCM corresponding to a cell density of 1 x 106 cells/mL (step 1.5.1), or the volume of FBS corresponding to a cell density of 6 x 106 cells per 0.9 mL of FBS (step 1.6.3).
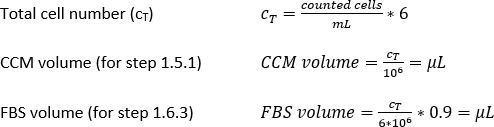
- Close the lid, place the tube in the centrifuge, and centrifuge as done in step 1.2.13. Remove and discard the supernatant without disturbing the cell pellet. Proceed to step 1.5 for cell seeding or step 1.6 for cell freezing.
- PBM seeding and differentiation into MDMs and MDDCs
- Resuspend the cell pellet in the calculated volume of CCM as calculated in step 1.4.2 (here, a final concentration of 1 x 106 cells/mL) by pipetting up and down 3x.
- Pipette the number of cells intended to differentiate into MDMs and MDDCs in separate conical centrifuge tubes using a serological pipette.
- Pipette differentiating factors to the CCM with PBMs and mix well by pipetting up and down. Differentiating factors are applied as follows:
- For MDDCs: final concentration of 10 ng/mL interleukin-4 (IL-4) and 10 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF).
- For MDMs: final concentration of 10 ng/mL macrophage colony-stimulating factor (M-CSF).
- Pipette the cell suspensions in CCM with the added differentiating factors into 6 well plates by distributing 3 mL of the suspension per well (corresponds to 3 x 106 cells/well, i.e., 1 x 106 cells/mL).
- Place the 6 well plates in a cell culture incubator (37 °C, 5% CO2) and let them differentiate for 6 days without refreshing the CCM.
NOTE: Differentiation ranges from 5–8 days depending on the local availability of buffy coats and experimental setup, provided that the differentiation efficiency is determined using the appropriate techniques (refer to the discussion section).
- PBM freezing
- Resuspend the cell pellet in a cryoprotective medium (here, FBS and dimethyl sulfoxide [DMSO; cytotoxic]) at a ratio of 9:1 (v/v) by pipetting a volume of prewarmed FBS. This corresponds to a final cell concentration of 6 x 106 cells/mL, considering a further addition of 10% DMSO (v/v).
- Mark the desired number of cryovials in the laminar flow hood (i.e., record the date, isolation code, and number of cells).
- Pipette 0.9 mL of cell suspension in pure FBS (here, 6 x 106 cells in 0.9 mL of FBS) to each cryovial. Subsequently, slowly pipette 0.1 mL of DMSO and mix the suspension well by turning the cryovials up and down 3x.
- Transfer the cryovials to a cell-freezing container and immediately set it to -80 °C for 24 h.
- After 24 h, remove the cryovials from the -80 °C freezer and container and place them into the liquid nitrogen tank suitable for cell storage.
- PBM thawing and differentiation into MDMs and MDDCs
- Warm all the required reagents to 37 °C in a water bath (~20–30 min).
- Prepare the appropriate number of 6 well plates corresponding to the number of thawed cells (here, one plate per 1.8 x 107 cells, i.e. 3 cryovials). Pipette 2 mL of CCM to each well under aseptic conditions. Place plates in the incubator (5 % CO2, 37 °C) for 15 min to allow the pH to equilibrate.
- Take the required amount of cryovials with frozen cells from a liquid nitrogen tank and gently swirl them in a 37 °C water bath (1–2 min) to ensure uniform thawing of the cell suspension.
- Remove the cryovial from the water bath and decontaminate with a sterilizing agent, ensuring that the agent does not interact with the lid and O-ring.
NOTE: Hereon, all steps must be completed under aseptic conditions. - Prepare the appropriate number of 15 mL conical centrifuge tubes corresponding to the number of cryovials to be thawed (here, 6 x 106 cells/tube). Pipette 9 mL of prewarmed CCM into each tube.
- Pipette slowly (drop-by-drop) the contents of the cryovial into a tube containing CCM. Close the lid, repeat for each tube, and centrifuge at 200 x g for 5 min at a regular centrifuge speed.
- Discard the supernatant without disturbing the pellet.
- Resuspend the pellet from each tube (which contains cells from one cryovial) in 2 mL of prewarmed CCM by pipetting up and down using a serological pipette (cell density corresponding to 3 x 106 cells/mL).
- From each tube, pipette the resuspended cells into two wells (1 mL per well) of a 6 well plate containing 2 mL of previously prepared CCM to reach a cell density of 3 x 106 cells/well (corresponding to a final concentration of 1 x 106 cells/mL). Repeat this for all other tubes.
- Proceed with differentiation as described in section 1.5.3.
- Place the 6 well plates in a cell culture incubator (37 °C, 5% CO2) and let them differentiate for 6 days without refreshing the CCM.
2. Triple cell coculture model of human alveolar epithelial tissue
NOTE: This section provides instructions on volumes and cell numbers corresponding to 12 well plate inserts. Figure 1 summarizes a proposed timeline for model assembly.
- Epithelial cell (A549 cell line) seeding
- Culture the epithelial cells according to the recommendations provided by the supplier (ATTC). Briefly, subculture the cells in CCM at 80% cell confluency (approximately, 2x–3x per week).
NOTE: Subculture A549 for at least four passages before the coculture model composition, using A549 cells in a passage range of 5–25. - Pipette 1.5 mL of prewarmed CCM into 12 well plates (the number of wells corresponds to the desired number of models).
- Place individual 12 well cell culture inserts into wells of a 12 well plate using sterilized tweezers.
- Detach the cells from a flask according to the subcultivation protocol (i.e., using a detaching agent, remove the agent by centrifugation as done in step 1.2.13). Resuspend in the appropriate volume of CCM corresponding to the final cell concentration of A549 (here, 50 x 104 cells/mL; 0.5 mL of cell suspension per insert, i.e. 25 x 104 cells/insert, which corresponds to seeding density of 27.8 x 104 cells/cm2).
- Pipette 0.5 mL of the cell suspension (i.e., 25 x 104 cells/insert) into the apical side of the insert using a 1 mL pipette.
- Cover the plates with lids and place them in a cell culture incubator (37 °C, 5 % CO2) for 4 days.
NOTE: Regularly check the confluency of A549 cells under a phase-contrast microscope.
- Culture the epithelial cells according to the recommendations provided by the supplier (ATTC). Briefly, subculture the cells in CCM at 80% cell confluency (approximately, 2x–3x per week).
- MDDC seeding
- Aspirate CCM with unattached cells in the 6 well plates containing MDDCs.
- Add 1 mL of fresh prewarmed CCM to each well.
- Use a cell scraper, detach (scrape) adherent MDDCs from each well, gently wash the wells with the existing 1 mL of CCM 3x, and combine them into one conical centrifuge tube.
- Count the cells with a cell counter using the trypan blue exclusion method using 10 μL of cell suspension and 10 μL of trypan blue solution.
- Centrifuge the cell suspension as done in step 1.2.13.
- Calculate the CCM volume required for resuspension (Equation 5):
<!–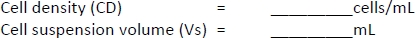
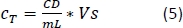
–> Required MDDC density = 42 x 104 cells/mL; each insert requires 6.3 x 104 cells, which corresponds to a seeded cell density of 7 x 104 cell/cm2 (here, 150 µL added onto 0.9 cm2 in the step 2.2.10.).
CCM volume for cell resuspension (Vm; Equation 5):
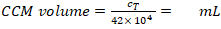
- Gently aspirate and discard the CCM from the upper chamber of 12 well plates with growing A549 on the inserts.
- Place the inserts with A549 cells in an upside-down position in a sterile Petri dish using sterilized tweezers. Prepare a conical centrifuge tube (50 mL) with PBS and pre-moisten a cell scraper.
- Scrape off A549 cells from the basal surface of the insert (i.e., the upper part at the upside-down position), which should grow through the pores of the inserts.
NOTE: Rinse the scraper with PBS (prepared in a tube) between scraping individual samples and keep it wet throughout the procedure. - Upon centrifugation (step 2.2.5), aspirate and discard the supernatant, then redisperse the MDDC pellet in the calculated amount of CCM (step 2.2.6) and pipette up and down 3x.
- Pipette 150 µL of the cell suspension on top of each insert so that the entire basal surface of the insert is equally covered with the liquid and not containing bubbles.
- Cover the dish with a lid and place in a cell culture incubator for 70 min. Aspirate the CCM from the cell culture plates (where the inserts have initially been placed), discard it in a biohazard liquid waste, and pipette 1.5 mL of fresh CCM into each well. Cover the plate with a lid and place in the cell incubator (37 °C, 5% CO2).
NOTE: Do not exceed the time period mentioned above to avoid drying out the cells. - After the incubation, carefully hold each insert with sterilized tweezers and place them to the plates containing CCM in an ordinary position. Cover the plate with a lid and return it to the cell incubator (37°C, 5% CO2).
- Macrophage (MDM) seeding
- Take 6 well plates containing predifferentiated MDMs. Place them from the cell incubator to a laminar flow hood.
- Aspirate and discard CCM with unattached MDMs grown in 6 well plates, and pipette 1 mL of fresh prewarmed CCM in each well.
- Using a cell scraper, gently remove adherent MDMs from individual wells (as done in step 2.2.3 for the MDDCs).
- Pipette 10 µL of trypan blue into a well or tube and add 10 µL of the MDM suspension to achieve the final dilution of 1:1 (v/v). Count the number of MDMs using the appropriate counting protocol.
- Centrifuge the cell suspension as done in step 1.2.13.
- Calculate the required volume (Equation 6):
Required MDM density = 2.5 x 104 cells/mL in CCM (here, each insert requires 1.25 x 104 cells in 0.5 mL CCM, which corresponds to a seeded cell density of 1.4 x 104 cell/cm2).
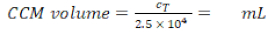 (6)
(6) - Upon centrifugation, aspirate and discard the supernatant, redisperse the MDM pellet in the calculated amount of CCM (step 2.3.6), and pipette up and down 3x.
- Carefully pipette 0.5 mL of the MDM suspension (prepared in step 2.3.7) on the wall of the cell culture inserts with A549 and MDDCs (not directly on epithelial cells) using a 1 mL pipette. Cover plates with lids and place in a cell culture incubator (37 °C, 5 % CO2) for 24 h.
- Transfer of coculture model to air-liquid interface (ALI)
- Upon the end of the 24 h (±2 h) incubation period of the assembled model in a cell culture incubator, aspirate and discard CCM from both apical and basal parts of cell culture inserts and from the wells.
- Using sterilized tweezers, lift individual inserts from the wells and pipette 0.6 mL of fresh prewarmed CCM to each well using a 1 mL pipette. Do not add CCM to the apical side of the insert.
- Cover plates with lids and place in a cell culture incubator (37 °C, 5 % CO2) for 24 h prior further use.
3. Exposure to selected positive controls (known stimuli for inducing proinflammatory response)
NOTE: Exposure of the coculture models to a known proinflammatory stimulus endotoxin lipopolysaccharide (LPS)7 and proinflammatory cytokine tumor necrosis factor α (TNF-α)7 is used to illustrate responsiveness of the model. Furthermore, exposure to a detergent (Triton X-100) is used to confirm the sensitivity of a lactate dehydrogenase (LDH) assay.
- Prepare the positive control solutions: LPS stock (1 mg/mL in distilled water), TNF-α stock (100 μg/mL in distilled water) and Triton-X 100 (2% [v/v] in PBS).
- Upon 24 h incubation of the coculture model at ALI conditions, aspirate and discard the supernatant from the basal compartment. Using sterilized tweezers, lift individual inserts from the wells and pipette 0.6 mL of fresh prewarmed CCM into each well.
- Prepare individual positive controls working solutions by diluting the stocks in CCM in conical centrifuge tubes as follows: 1 μg/mL LPS, 1 μg/mL TNF-α, and 0.2% Triton-X 100. The volumes correspond to the number of tested inserts (here, 100 μL/insert). Mix the solutions well by pipetting up and down 3x.
- Apply 100 μL of each positive control solution by slowly pipetting onto the wall of the cell culture insert. Cover the well plate with a lid and place in the cell culture incubator (37 °C, 5% CO2) for 24 h. Upon incubation, aspirate and discard the liquid at the apical side of the insert by holding individual inserts using tweezers.
- Collect CCM in the basal compartments and store at 1) 4 °C for further LDH analysis, denoting cell membrane rupture-mediated cytotoxicity, and/or 2) store at -80 °C for further analysis of protein release via enzyme-linked immunosorbent assay (ELISA). Run the assays according to the kit supplier recommendations.
- Upon the removal of CCM, wash the inserts with PBS 3x and fix the cells on cell culture inserts in 4% [w/v] paraformaldehyde (in PBS, 15 min at room temperature) by ensuring both insert sides are well-covered with PFA solution. Subsequently wash 3x with PBS to remove PFA. Store the samples submerged in PBS at 4 °C for further immunostaining (an example of this method has been described previously17).
Human lung coculture models, composed of alveolar epithelial cells and immune cells, were assembled either from fresh or frozen MDDCs and MDMs progenitors (here, human peripheral blood-derived monocytes). As presented in Figure 1, A549 cells were seeded 3 days after the first section involving monocyte isolation/thawing. After 6 days of differentiation, the differentiated MDMs appeared round-shaped, whereas MDDCs formed a more elongated shape with observable protrusions. They also appeared as agglomerates, especially when differentiated from fresh monocytes (Figure 2, Figure 3). Epithelial cells formed a dense cell layer of cells after 3 days of growth on membrane inserts (Figure 4), when the cocultures were assembled. After 24 h of assembling and an additional 24 h of being subjected to ALI conditions, the cocultures were prepared for exposures.
Responsiveness of the 3D cell culture models was investigated upon exposure to known proinflammatory stimuli using a pseudo-ALI approach, as described previously29. The proinflammatory stimuli, LPS and TNF-α, were added in low volumes (100 μL) onto the apical surface of the air-exposed cell model. In parallel, the absence of membrane rupture as a measure of cytotoxicity was assessed via LDH assay. A significant increase in LDH release in CCM of the basal compartment was observed upon exposure to the positive control for membrane rupture, a detergent Triton-X 100 (Figure 5). These results proved responsiveness of the model to a cytotoxic substance, whereas no increase in LDH release was observed upon apical stimulation with TNF-α or LPS.
A possible reason for the different measured values of LDH in the samples assembled with either fresh or previously frozen PBMs can be attributed to the sample storage. Samples from fresh PBMs were stored for a longer time at -80 °C; therefore, the activity of LDH enzyme may drop. Notably, LDH is stable for only up to 4 days in CCM; thus, it is recommended to perform the assay the latest 2 days after collecting the supernatants. Alternatively, it is possible to freeze down the supernatants directly after collection. However, it is important to consider that freezing can decrease the enzymatic activity of LDH.
Secretion of proinflammatory mediators (here, TNF-α and interleukins 6 [IL-6] and 8 [IL-8]) into the basal CCM was quantified via ELISA. Statistically significant (p < 0.05, one-way ANOVA) increases in the release of IL-6 and IL-8 were observed in both LPS- and TNF-α- treated samples compared to respective untreated cells, as well as in the cell culture models assembled from either PBMs source (Figure 6). Although concentrations (pg/mL) of all the tested cytokines in the basal CCM were higher in the cocultures composed of fresh PBMs, differences among the two cocultures and monocultures were not statistically significant (p > 0.05) (Figure 6). To confirm the added value of coculture models with respect to a 2D epithelial cell culture, A549 monocultures were also exposed to LPS or TNF-α. As expected, the release of all investigated mediators from A549 monocultures was lower compared to both coculture models; although, the difference between them was not statistically significant (p > 0.05, one-way ANOVA).
Cellular morphology of the 3D human alveolar epithelial tissue barrier was assessed via confocal laser scanning microscopy (LSM). To visualize the composition of each model, macrophages within the coculture models (MDMs) were stained with mature macrophage marker 25F9. MDDCs were stained with CD83, which is an important marker for activated dendritic cells30. Regarding cellular morphology, no difference was observed between the coculture models using MDMs and MDDCs from fresh PBMs compared to those using thawed PBMs. In LPS- and TNF-α-exposed cocultures, both composed of fresh and frozen immune cells, a disrupted epithelial layer in LSM images was observed, which was not the case in untreated cells (Figure 7, Figure 8).
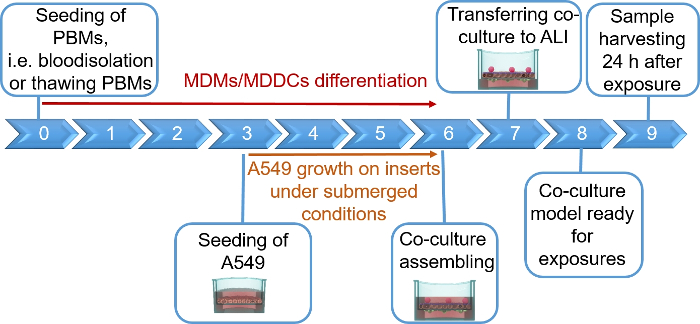
Figure 1: Schematic timeline of the protocol. Presentation of the 3D coculture model preparation, assembly, and application (exposure to a tested substance). ALI = air-liquid interface, MDDCs = monocyte-derived dendritic cells, MDMs = monocyte-derived macrophages, PBMs = peripheral blood monocytes. Please click here to view a larger version of this figure.
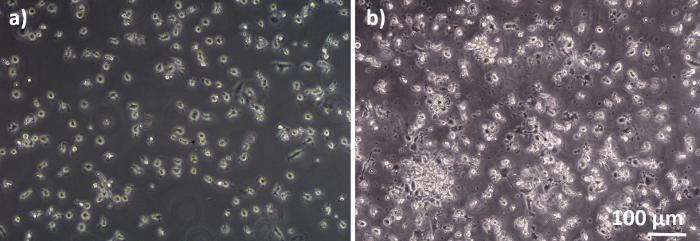
Figure 2: MDMs and MDDCs differentiated from fresh PBMs. Phase-contrast microscopy image of differentiated (A) MDMs and (B) MDDCs from fresh PBMs (6 days after cell isolation). MDMs are round-shaped, while MDDCs are often observed as agglomerates. Please click here to view a larger version of this figure.
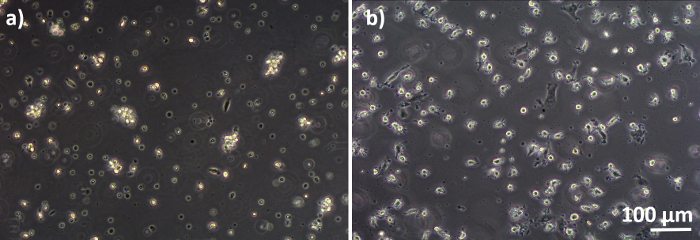
Figure 3: MDMs and MDDCs differentiated from frozen PBMs. Phase-contrast microscopy image of differentiated (A) MDMs and (B) MDDCs from thawed PBMs (6 days after thawing). MDMs are round-shaped, but some elongated cells can be observed. MDDCs also appear round-shaped with protrusions. Please click here to view a larger version of this figure.

Figure 4: Epithelial cells growth on membrane inserts. Phase-contrast microscopy image of confluent A549 growing on a membrane insert 4 days after seeding, forming a dense layer of cells. Please click here to view a larger version of this figure.
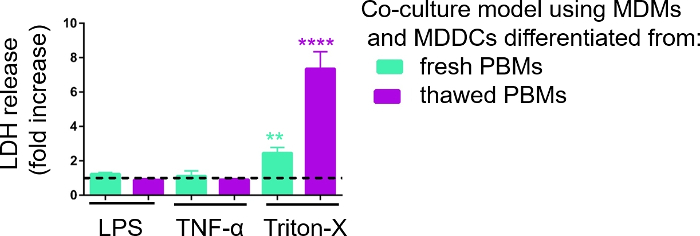
Figure 5: Cytotoxicity results investigated via membrane rupture-based (LDH) assay. The data is presented as a fold increase over untreated cells (mean ± SD, n = 3, asterisk denotes statistically significant increase compared to untreated cells, **p < 0.01, ****p < 0.0001). In green models MDMs and MDDCs from fresh PBMs are represented, and in purple models assembled from thawed PBMs are represented. Please click here to view a larger version of this figure.
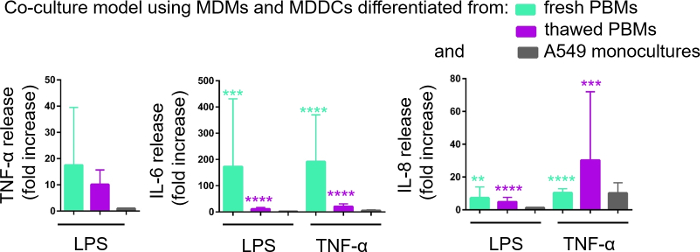
Figure 6: Proinflammatory reactions in the cocultures and monocultures. The proinflammatory mediators (TNF-α, IL-6, and IL-8) release in the cocultures upon 24 h challenge with LPS or TNF-α. The data is presented as relative to untreated cells (mean ± SD, n = 3, **p < 0.01, ***p < 0.001, ****p < 0.0001). In green models MDMs and MDDCs from fresh PBMs are represented, and in purple models assembled from thawed PBMs are represented. Grey represents A549 monocultures. Please click here to view a larger version of this figure.
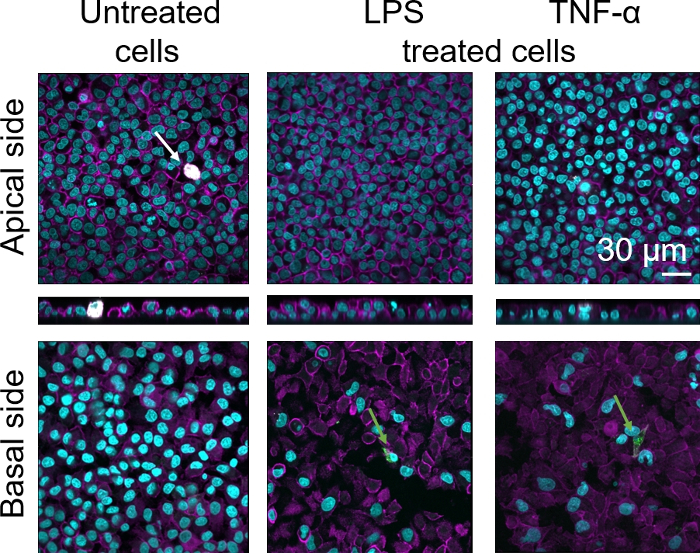
Figure 7: Morphology of cocultures composed of fresh immune cells. LSM images of apical and basal sides of the coculture model with xz projections of apical sides of the model using MDMs and MDDCs from fresh PBMs. Cyan represents nuclei (DAPI), magenta represents cytoskeleton (rhodamine-phalloidin), white represents MDMs (25F9), and green represents MDDCs (CD 83). The white arrow denotes MDM, whereas the green arrow denotes MDDC. Please click here to view a larger version of this figure.
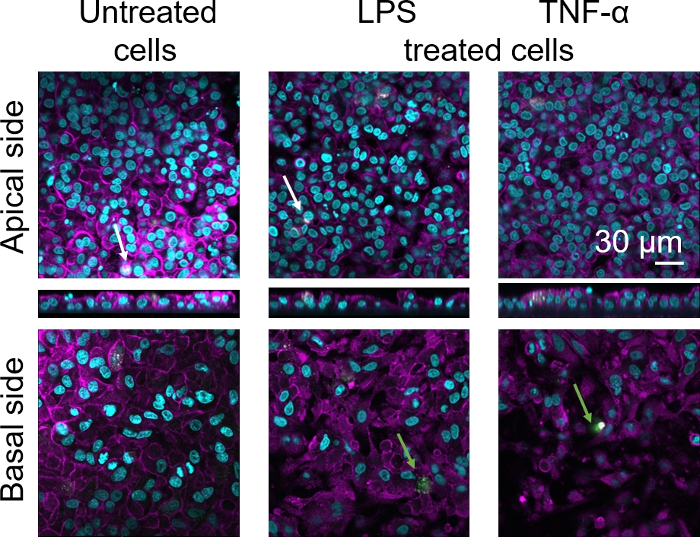
Figure 8: Morphology of cocultures composed of frozen immune cells. LSM images of the apical side of the coculture model with corresponding xz projections, and basal side of the model using MDMs and MDDCs from thawed PBMs. Cyan represents nuclei (DAPI), magenta represents cytoskeleton (rhodamine-phalloidin), white represents MDMs (25F9), and green represents MDDCs (CD 83). The white arrow denotes MDM, whereas the green arrow denotes MDDC. Please click here to view a larger version of this figure.
