This method of DNA halo preparation has helped us in our endeavors to determine differences in genome behavior within young and old cells, but also in cells derived from premature ageing diseases with aberrant nucleoskeletal proteins15. Figure 1 displays examples of DNA halos where it is possible to see the edge of a residual nucleus, the DNA remaining within the residual nucleus and the unattached DNA that has spooled out into the surrounding area creating a DNA halo. It also depicts the analysis showing how the residual nucleus is obtained and the NE and CTE measurements. It is possible to differentiate between proliferating and non-proliferating cells by either incorporating a labeled nucleotide such as BrdU when cells are in S-phase or employing the diagnostic proliferation marker anti-pKi67, which reveals nucleoli, and regions of heterochromatin in G1 cells17,18. Primary cells grown in high serum without achieving confluency, that are negative for the proliferation markers, are assumed to be senescent. Primary cells grown in low serum or have become confluent i.e., contact inhibited that are negative for the proliferation markers are deemed quiescent and would be able to reenter the proliferative cell cycle given the correct nutrients and situation. Being able to differentiate between Ki67 positive and negative cells has enabled us to determine differences between proliferating, quiescent and senescent human dermal fibroblasts. Figure 2 displays DNA halos of proliferating human dermal fibroblasts created from cells where BrdU was incorporated into them during DNA replication, a mechanism that does not occur in non-proliferating cells, and subsequently stained with anti-BrdU antibody. Staining with the proliferative marker anti-pKi67 antibody is also visible in Figure 2. This is a robust antigen and survives the FISH protocol and so can be stained for post-FISH and pre-mounting. Thus, proliferating cells are positive (red) for BrdU and anti-pKi67 (red) in the left-hand column and non-proliferating cells, indeed senescent cells in Figure 2 are displayed in the right-hand column. The green signals are individual telomeres revealed with a telomere PNA FISH/FITC kit. Combining immunofluorescence with DNA halos enables analysis during different cell states, as shown in Figure 2 when investigating proliferating, quiescent and senescent cells. Depending on the antibody chosen other conditions can be examined, such as differentiation, DNA damage via irradiation etc.
Chromosome territories can also be visualized within DNA halos using FISH. Due to the preparation permitting spooling of DNA out the nuclei, the chromosome territory shape can be disturbed, with smaller or larger amounts of the chromosome found in the DNA halo, depending on the anchorage of the genome inside the residual nucleus and its structures. Figure 3 reveals a panel of DNA halos whereby individual chromosomes have been revealed with specific whole arm chromosome painting probes (red) for chromosomes 1, 13, 17 and 18. Anti-pKi67 (green) has been used to mark proliferating cells and its absence within the same culture, upon the same slide, denoting senescent cells. It is very obvious from the images and the data presented as CTE/NE that the small gene-poor chromosome 18 is a chromosome that has few attachments and spools further out into the DNA halo away from the residual nuclei and is significantly further from the center of the residual nuclei than the other chromosomes. However, this is also true for chromosome 1 as well. Using the proliferative marker anti-pKi67 it has also been possible to compare proliferating with senescent cells, within the same culture, and on the same slide, and this analysis has revealed that chromosomes within these two very different cell statuses are not significantly different from one another, with respect to attachment with the residual nuclear structures.
Interestingly, genes also are showing statistically significant differences between proliferating and senescent cells with respect to remaining within a residual nucleus or being located in the DNA Halo. Figure 4 demonstrates this with gene loci delineated by labeled BAC probes in red and anti-Ki67 in green. There are no significant differences between gene locations in the proliferating versus the senescent cells, after a DNA Halo preparation. However, there are significantly more catenin alpha 1 CTNNA1 loci within the DNA halo than cyclin D1 CNDD1 loci, where there are very few. Figure 5 displays DNA halo preparations with telomeres in green. The background is left deliberately high to enable telomere signals to be visualized within the DNA halo. In this set of data quiescent cells i.e., cells that have been serum starved for 7 days have been included and interestingly there are significantly more telomeres unattached and located within the DNA halos in quiescent cells than for proliferating and senescent cells. In Figure 5a the proportion of telomeres in the DNA halo can be observed, particularly for the image 'Experiment 2'. This corresponds with Figure 5b where the mean percentage of telomeres in DNA halo is approximately 17% in quiescent cells. There is some evidence that not all telomeres in senescent cells can be seen as some of them maybe very short.
This method of DNA halo has been successful for us to investigate genome interaction alterations within nuclei in diseased cells15. Figure 6 demonstrates differences in chromosome attachment in primary control fibroblasts and in diseased cells with typical (lamin A mutation) and atypical Hutchinson-Gilford Progeria Syndrome, expressing a different SUN1 isoform and no lamin A mutation19. Chromosomes 1 and 13 show statistically significant differences in their attachment within the residual nuclei when compared to control DNA halos. Figure 6b correlates the position of the whole chromosome territory to the residual nucleus and DNA Halo. Values of 1 or less indicates the chromosome is located within the residual nucleus and values over 1 demonstrate chromosomes or portions of chromosomes within the DNA Halo.
Overall, this highlights the utility of HALO-FISH in investigating genomic interactions of whole chromosomes, specific genes and telomeres under a variety of conditions that affect the cell cycle (proliferation, quiescence and senescence) or within disease cells e.g., progeria and cancer cell lines. Indeed, the differences in interactions between these states implies the nucleoskeleton has an important role in regulating key processes within the nucleus.
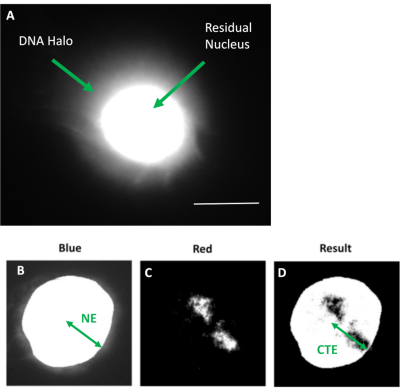
Figure 1: HDF extracted nucleus displaying the residual nucleus and DNA halo and overview of analysis method. (a) An HDF nucleus prepared via DNA halo assay and counterstained with DAPI. The brightly stained residual nucleus shows DNA anchored to the nucleoskeleton and this is surrounded by the non-attached DNA which forms a halo of DNA. Magnification = x 100; scale bar 10 µm. (b) The blue channel captures the DAPI-stained nucleus and surrounding DNA. The residual nucleus is selected and removed using ImageJ. The arrow depicts the distance from the nuclear center to the residual nuclear edge (NE). (c) The red channel shows the probe signal. (d) The image denoted 'Result' is the outcome of superimposing the red channel on the blue channel image; this allows the distance from the nuclear center to the furthest chromosome territory edge (CTE). Please click here to view a larger version of this figure.
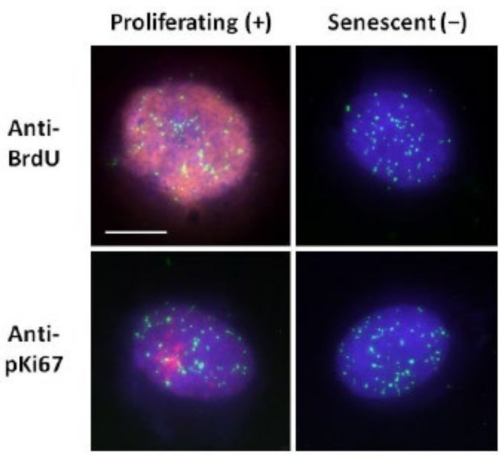
Figure 2: DNA halo preparation with telomere PNA FISH on proliferating and senescent HDFs. Telomere PNA FISH on HDFs subjected to DNA halo assay. Telomere signals are visualized in green (FITC), residual and halo DNA was counterstained using DAPI (blue) and proliferating nuclei were detected using either anti-BrdU or anti-pKi67 antibodies via indirect immunofluorescence in red (TRITC). Magnification = x 100; scale bar 10 µm. Please click here to view a larger version of this figure.
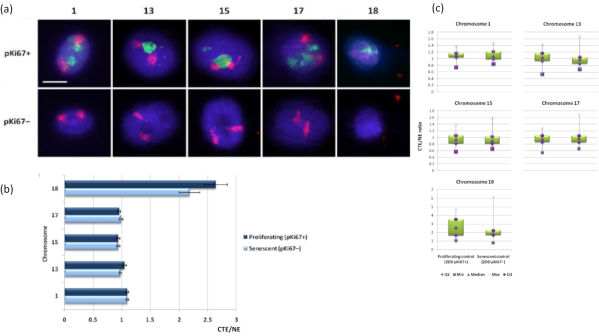
Figure 3: Nucleoskeleton-chromosome interactions and analysis using DNA halo assay. (a) 2D-FISH with probes specific for chromosomes 1, 13, 15, 17 and 18 was performed on HDFs subjected to DNA halo preparation. Whole chromosomes were painted in red (Cy3) and nuclei were probed with pKi67 to determine if they were proliferating or senescent. Proliferating cells (pKi67+) were delineated in green (FITC), whereas senescent cells remained unstained (pKi67-) i.e. no green signal detected. Magnification = x 100; scale bar 10 µm. (b) Chromosome anchorage by the nucleoskeleton in proliferating and senescent HDFs that had undergone HALO-FISH. Measurements show the ratio of the furthest chromosome territory edge (CTE) to respective nuclear edge (NE) for chromosomes 1, 13, 15, 17 and 18 in proliferating (pKi67+) and senescent (pKi67-) cells. Error bars represent ± SEM. (c) Modified box plot representation of chromosome territory edge (CTE) to respective nuclear edge (NE) of specific chromosomes in pKi67+ and pKi67- nuclei. Q1 = lower quartile; Min = lowest value recorded; Med = median; Max = maximum value recorded; Q3 = upper quartile. Please click here to view a larger version of this figure.
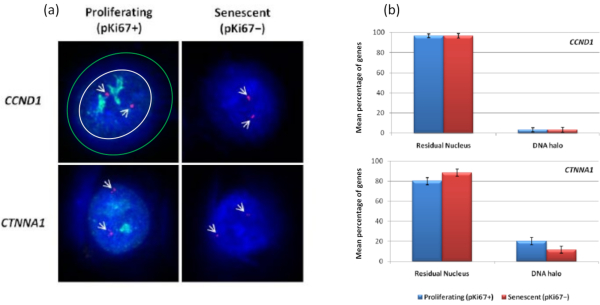
Figure 4: Gene-specific interactions in HDFs using HALO-FISH. (a) DNA halo extracted nuclei were probed with gene specific probes (CCND1 and CTNNA1) to investigate their anchorage to the NM on proliferating and senescent cells. The gene signals are shown in red (Cy3) and anti-pKi67 depicts proliferating cells and signal is visualized in green (FITC). For the proliferating CCND1 image, the residual nucleus is enclosed within the white circle, and the space between the white and green circle depicts the DNA Halo. Magnification = x 100; scale bar 10 µm. (b) Gene-specific signals for CCND1 and CTNNA1 are compared between the residual nucleus and DNA halo, and also, between proliferating and senescent cells. Error bars represent ± SEM. Please click here to view a larger version of this figure.
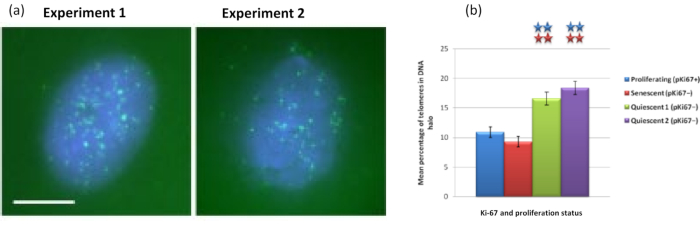
Figure 5: DNA halo assay on quiescent HDFs probed with telomere PNA-FISH. (a) Quiescence of HDFs was induced by culture in low serum medium for 7 days. The DNA halo assay was performed, and PNA-FISH enabled visualization of telomeres by FITC signal (green) and the residual nucleus and surrounding DNA halo was counterstained with DAPI (blue). Cells were also stained with anti-pKi67 antibody to ensure nuclei were non-proliferating. This was repeated on two separate occasions. Magnification = x 100; scale bar 10 µm. (b) Comparison of the mean percentage of telomeres localized within the DNA halo in proliferating, senescent and quiescent HDF cells. Error bars represent ± SEM. Please click here to view a larger version of this figure.
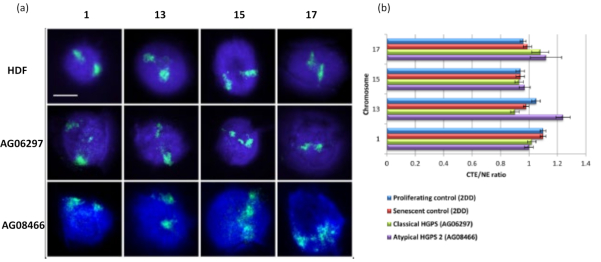
Figure 6: Examining whole chromosome anchorage to the nucleoskeleton in HGPS cells using HALO-FISH26. (a) Control HDF (2DD), classical HGPS (AG06297) and atypical type 2 HGPS (AG08466) nuclei underwent DNA halo preparation and then 2D-FISH using whole chromosome paints for chromosome 1, 13, 15 and 17. Whole chromosomes are depicted in green (FITC) and DNA was counterstained with DAPI (blue). Magnification = x 100; scale bar 10 µm. (b) Positioning of chromosomes within extracted nuclei was determined by measuring the ratio of the mean chromosome territory edge (CTE) to the nuclear edge (NE). A ratio above 1 demonstrates that the furthest CTE lies outside the corresponding NE within the DNA halo, while a ratio below 1 signifies that the furthest CTE lies within the NE within the residual nucleus. Please click here to view a larger version of this figure.
| Constituents | Volume(μL) |
| 5XDOP-PCRbuffer | 10 |
| dNTPmix(withoutdTTP)(2mM) | 5 |
| dTTP(2mM) | 2 |
| Biotin-16-dUTPorDigoxigenin-11-dUTP | 10 |
| DOPprimer(20μM) | 5 |
| TaqDNAPolymerase(1U/μL) | 1 |
| PCRgradewater | 12 |
| Template | 5 |
Table 1: Table showing the DOP-PCR components and volumes for a 1x reaction
| Step | Cycles | Temp (degree Centigrade) | Time |
| Initial Denaturation | 1 | 95 | 3 min |
| Denaturation | 34 | 98 | 20 s |
| Primer Annealing | 62 | 1 min | |
| Extension | 72 | 30 s | |
| Final Extension | 1 | 72 | 5 min |
| Cooling | 4 | Hold |
Table 2: Table showing the DOP-PCR cycle, temperature, and time profile.
| Constituent | Volume(μL) |
| 10x NT buffer (0.5M Tris-HCl pH 8,50 mM MgCl2, 0.5 mg/ml BSA) | 5 |
| 0.1 M beta-mercaptoethanol | 5 |
| 10X Nucleotide stock (0.5 mM dATP, 0.5 mM dCTP, 0.5 mM dGTP, 0.5 mM dTTP, 0.5 mg/ml biotin-16-dUTP) | 5 |
| Dnase I (1 ng/ml) | 2 |
| DNApolymerase I | 5U per μg of DNA |
| DNAtemplate (1 μg) | 1 |
| DEPC-treated water | To 50 μL |
Table 3: Table showing the nick translation components and volumes for a one probe.
