All experiments involving animals were carried out with a protocol approved by the KUMC Institutional Animal Care and Use Committee, in accordance with their guidelines and regulations (Protocol Number: 2018-2447).
1. Harvest E13.5 embryos
- Euthanize pregnant female mice using a CO2 inhalation chamber or by a procedure approved by the Institutional Animal Care and Use Committee. Immediately proceed to dissection.
- Expose the inferior half of the abdominal cavity by removing the skin and peritoneum. Excise both horns of the uterus, which contain the E13.5 embryos.
- Briefly place the uterus in prewarmed 37 °C sterile phosphate-buffered saline (PBS) to rinse off excess blood, hair, or other debris. Place the uterus in a sterile 10 cm dish filled with sterile PBS.
- Using small scissors, cut through the uterine wall along the length of the uterus to expose each embryo, still in its yolk sac. Remove the yolk sac surrounding the embryo, but save it for genotyping, if needed. As the embryos are removed, place each embryo in its own well of a 12-well plate filled with PBS.
2. Dissection of palatal shelves from embryos (Figure 1)
NOTE: Sterilize the stainless steel dissection instruments (see the Table of Materials) after processing each embryo by placing the instruments first in a beaker of 100% ethyl alcohol (EtOH), then in an instrument sterilizer at 350 °C for 10 s, and then cooling them in a second beaker of 100% EtOH.
- Using a sterilized perforated spoon, place the embryo in a new 10 cm dish filled with MEPM culture medium consisting of Dulbecco's minimum essential medium (DMEM) containing 10% fetal bovine serum (FBS), L-glutamine (4 mM L-Glu), and the antibiotics-penicillin and streptomycin (50 units/mL).
- Decapitate the embryo right below the jaw line using sterile scissors (Figure 1A, red dotted line). Remove the lower jaw by inserting one point of the sterilized fine #5 forceps into the mouth, keeping it just inside the cheek. Push the point of the inserted forceps through until it exits out the back of skull.
- Orient the forceps, along the yellow line in Figure 1B, so that the other side of the forceps (which is still outside of the embryo) is hovering just over the ear canal, then pinch the forceps shut to cut the tissue. If necessary, run another fine forceps along the seam of the now closed forceps to cut through any tissue that was not completely severed by the pinch.
- Repeat the previous step for the other side of the embryo head. Continue the pinch-cut procedure to fully remove the lower jaw, tongue, and inferior portion of the skull and expose the palatal shelves.
- Remove the cranium of the skull by cutting just above the eyes, as shown in Figure 1C (green line). Do this by placing the head on its left or right side and positioning the points of the small stainless steel scissors in front of and behind the skull just above the embryo's eye level. Cut off the top of the skull with one fast snip of the scissors, creating a flat surface that will be important for stability in later steps and that should look like Figure 1D when viewed from the side.
- Place the remaining part of the head upside down, with the superior aspect of the head (cranium removed) resting flat on the bottom of the dish, which will provide a stable surface for palatal shelf removal. Take a moment to identify the palatal shelves, which are now exposed and facing up and will appear as two raised ridges on either side of a central groove in the anterior half of the head (Figure 1E).
- Pin the remaining portion of the head to the dish to immobilize it while the shelves are removed. Do this by inserting one point of a fine forceps through the tissue near the nasal region of head, anterior to the palatal shelves, and insert the other point of the forceps through the base of the skull, posterior to the palatal shelves. Hold these in place while performing the excision of the palatal shelves.
- Immobilizing the head with one hand, pick any one of the two shelves to remove first, and insert both points of a second pair of fine forceps into the tissue at the base of the lateral surface of the shelf, and pinch to cut the tissue (Figure 1F). Repeat this along the base of the medial surface of the shelf and then at both the anterior and posterior ends of the shelf to detach from the head.
- Gently lift the shelf, making additional pinches, as needed, to completely free the shelf from the surrounding tissue.
- Repeat the previous two steps to remove the second palatal shelf.
- With the palatal shelves now freed from the surrounding tissue and placed in PBS (Figure G), use a sterile plastic bulb transfer-pipette to draw up the shelves in the pipette, and transfer them into a 1.5mL microcentrifuge tube along with approximately 500 µL of PBS. Keep the tubes containing palatal shelves on ice as the rest of the litter is processed in the same fashion.
NOTE: Alternatively, shelves can be placed in a 1.5mL microcentrifuge tube containing prewarmed trypsin (0.25%) immediately after dissection (in lieu of placing them on ice). Samples will be fresher, but care must be taken to time all the proceeding steps for each individual sample as opposed to treating the samples collectively.
3. Culture of MEPM cells
NOTE: Under the conditions described here, the palate epithelial cells do not survive the first passage, resulting in a pure palate mesenchymal cell culture. Use sterile technique to perform all steps in a tissue culture hood.
- Aspirate and discard the PBS from the 1.5 mL tube, taking care not to discard the shelves in the process. Immediately add 200 µL of prewarmed (37 °C) trypsin (0.25%) to each tube that contains palatal shelves. Briefly pipet the shelves up and down in the trypsin using a 1000 µL pipette tip to accelerate the trypsinization.
- Incubate the tubes for 5 min at 37 °C, then pipet each sample up and down again to help break up the tissue. Incubate the tubes for another 5 min at 37 °C, and pipet up and down once more to complete the dissociation of the tissue.
NOTE: The shelves must be completely or nearly completely dissociated and suspended in the trypsin with no visible chunks of tissue remaining. - Add 800 µL of MEPM culture medium (step 2.1) to each 1.5 mL tube. Centrifuge the 1.5 mL tube at 200 × g for 5 min to pellet the cells. Remove the supernatant, and resuspend the cell pellet in 1 mL of MEPM culture medium.
- Plate the MEPM cells into a 6-well tissue culture-treated plate containing MEPM culture medium. Allow the cells to adhere to the plastic surface for 12 h at 37 °C in an incubator with 5% CO2.
NOTE: After overnight incubation, the vast majority (~90%) of cells will attach. At this point, the adhered cells will look fairly homogeneous, with a triangular or slightly elongated shape. - Change the medium every day by gently aspirating the old medium and immediately replacing it with 1 mL of warm sterile PBS without calcium or magnesium for ~1 min. Aspirate the PBS, and replace with 3 mL of prewarmed MEPM culture medium.
- Passage the cells once they become 100% confluent.
NOTE: MEPM cells should proliferate by doubling in number almost daily.- To passage the cells, gently aspirate the old medium, and immediately replace it with warm PBS without calcium or magnesium for ~1 min. Aspirate the PBS, and replace it with 0.5 mL of prewarmed 0.25% trypsin.
- Incubate at 37 °C for ~5 min, or until the cells detach from the surface of the dish when gently rocked back and forth by hand. Once the cells have detached, immediately add 5 mL of prewarmed MEPM culture medium to the trypsinized cells.
- Using a 10 mL serological pipet, gently collect the cells in a 15 mL conical tube, and centrifuge the tube at 200 × g for 5 min to pellet the cells. Aspirate the trypsin and medium, and resuspend the cells in 3 mL of prewarmed MEPM culture medium. Gently pipet 1 mL of cells into a single well of a 6-well dish, and add 2 mL of MEPM culture medium to bring the total volume to 3 mL.
NOTE: This constitutes a 1:3 split of cells. MEPMs may be passaged up to three times. The seeding density of MEPMs is somewhat flexible, and the number of cells present varies depending on the culturing vessel. However, MEPMs do not properly proliferate when split too sparsely and should be at least 20-25% confluent in their new dish once they adhere.
4. Cryopreservation of MEPM cells
- Once trypsinized MEPM cells are pelleted, resuspend the cells in MEPM culture medium to obtain a concentration of ~ 1 × 106 cells/mL. Pipet the cells into cryovials, and add a final concentration of 5% dimethylsulfoxide in the cell stock. Cap the cryovial, briefly mix by inverting, and immediately place the vials in a freezing container that cools at a rate of 1 °C/min.
- Place the cooler in a -80 °C freezer overnight. On the next day, move the cryovials to a liquid nitrogen tank for long-term storage.
- Thawing cryopreserved MEPM cells
- Remove the cryovials from the liquid nitrogen tank, and thaw at room temperature until the contents begin to become liquid. Empty the contents into a 15 mL conical tube containing 9 mL of prewarmed MEPM culture medium.
- Centrifuge the tube at 200 × g to pellet the cells. Resuspend the cells in 1 mL of MEPM culture medium, and pipet the cells into a single well of a 6-well plate. Add 2 mL of warm MEPM culture medium to bring the total volume to 3 mL.
- Culture the cells at 37 °C in an incubator with 5% CO2. Change the medium daily.
5. Live-imaging of MEPM cells – 2D collective migration assay (Figure 2)
- Prepare a plate to use for live imaging.
- Use small surgical scissors or a sharp scalpel to shorten a sterile 2-well silicone insert to a height of ~1 mm. Prepare an insert for each sample being used.
- Using forceps, place the shortened 2-well silicone insert in the center of a well of a 6-well plate. Press down along all edges to ensure it is fully adhered.
- Thaw cryopreserved cells by following the steps in section 4.3 of this protocol. Count MEPM cells, and seed 300 cells/mm2 of the shortened silicone inserts in a total volume of 40-50 µL MEPM culture medium per well. Culture the cells overnight at 37 °C in an incubator with 5% CO2.
- On the next day, prepare for live time-lapse imaging.
- Use a phase contrast microscope with an on-stage incubator and automatic imaging capability. Add water to the onstage incubator reservoir to reduce evaporation of the culture medium; set the temperature to 37 °C and CO2 to 5%. Allow ~30 min for the humidity to build up before placing the 6-well dish in the onstage incubator.
- Use settings equivalent to the following for time-lapse imaging.
- Select the 4x objective to have large fields of view and phase contrast filter.
NOTE: Autofocus, Auto find sample, z-stack, and auto-lighting are not usually necessary. - Select two microscopic fields per well to capture the lumen of the shortened silicone inserts. Ensure all imaging positions have the correct focus.
NOTE: Image planes can be adjusted during imaging, but such adjustment is not usually necessary. - Select the desired image output file-type, then select or de-select post-imaging options, such as automatic video creation and watermarks, as desired. If applicable, select phase contrast as imaging mode.
NOTE: Using watermarks may impede subsequent image processing steps. - Set the duration of the recording to 72 h. Set the program to capture images every 10 min.
NOTE: Usually only 48 h of imaging is required, but imaging can be stopped at any point before the 72 h mark without losing images that have already been taken. - Make sure the environmental chamber is operational as required in 5.3.1. Save these settings (the routine) and begin imaging.
- Select the 4x objective to have large fields of view and phase contrast filter.
- Continue imaging until 72 h (or the specified time).
6. Live-imaging of MEPM Cells in a wound-repair assay (Figure 3)
- Prepare a plate to use for live-imaging. Using forceps, place a sterile 2-well silicone insert in the center of a well of a 6-well plate, and press down along all edges to ensure it is fully adhered. Prepare one 2-well insert for every sample being used.
- Thaw cryopreserved cells by following the steps in section 4.3 of this protocol. Count MEPM cells, and if necessary, concentrate the cells to at least 350 cells/µL.
- Seed 1400 cells/mm2 into the silicone inserts in a volume of 100 µL of MEPM culture medium per well. Culture the cells for 48 h at 37 °C in an incubator with 5% CO2, and change the medium every day.
- After 48 h prepare a microscope for live time-lapse imaging as described in sections 5.3 and 5.4. Immediately prior to placing the cells in the onstage incubator, add 3 mL of prewarmed MEPM culture medium to the well (but outside of the inserts), and then carefully remove the silicone inserts.
NOTE: The wall separating the 2 chambers leaves a gap that is the "wound". - Start time-lapse imaging as described in 5.4, with the following differences:
- Use a higher magnification (e.g., 10x) objective. To capture wound closure, select 5 fields of view along each wound, so that the wound is parallel with the vertical axis of the image.
- Stop imaging after 72 h or when the wounds have fully closed.
7. Computational analysis of time-lapse image sequences
NOTE: Perform the following procedures on a computer equipped with standard computational tools, such as the python interpreter, C compiler, and a shell (see the Table of Materials).
- Confluency analysis
NOTE: This procedure can be used to estimate cell proliferation within a sparse culture or to quantify wound closure experiments. To detect areas occupied by cells, a segmentation threshold is applied to the local standard deviation of image brightness. The code has been described previously by Wu et al.33 and Neufeld et al.34 and is available at http://github.com/aczirok/cellconfluency.- Determine the segmentation threshold for the images. As an example, to see the segmentation with a threshold 4, issue the command
segment.py -i inout-image.jpg -S 4 -test output.jpg
and then check the output (output.jpg).
NOTE: If the threshold is too low, background areas in the micrograph are classified as cell-covered. In contrast, if the threshold is too high, cell-covered areas are not classified as such. The optimal threshold value keeps both errors at a minimum. - Use the provided area.sh script to calculate confluency values for a sequence of images as
area.sh -S 4 img_001.jpg img_002.jpg …. > confluency.dat
NOTE: The discrimination threshold value 4 is verified in step 1, and the results are stored in the file confluency.dat. By sorting the images into appropriate folders, the list of image file names can be replaced by wildcard notation:
area.sh -S4 *.jpg > confluency.dat - For wound-closure experiments, transform the confluency data A(t)-the size of cell-covered area expressed as a percentage as a function of time-into wound-edge propagation speed V as
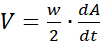
where w denotes the width of the field and dA/dt is the time derivative of A(t), i.e., the expansion rate of the cell-covered area.
- Determine the segmentation threshold for the images. As an example, to see the segmentation with a threshold 4, issue the command
- Cell motility map
- Execute a particle image velocimetry (PIV) algorithm to characterize cell motility, and extract the extent of local movement "optical flow" between image pairs, but not to identify individual cells.
NOTE: Here, an initial window size of 50 µm is used, as described in detail by Zamir et al.35 and Czirok et al.36, with an initial window size of 50 µm. The PIV analysis yields a velocity field v(x,t) for each image frame t and location (within the image) x. - Extract the average speed of cell motility from v(x,t) as a spatial average calculated over the cell occupied area, as determined in section 7.1.
- Execute a particle image velocimetry (PIV) algorithm to characterize cell motility, and extract the extent of local movement "optical flow" between image pairs, but not to identify individual cells.
- Manual cell tracking
NOTE: While the PIV analysis provides an automatic assessment of cell motility, to focus on the behavior of individual cells often requires manual tracking. While several tools provide this functionality, it is very helpful if the manually positioned markers can be modified after their initial placement, and if tracking can be performed both forward and backward in time.- Perform cell tracking with a custom-developed python tool (http://github.com/donnagreta/cm_track), which also provides basic editor functions such as deleting trajectory segments.
NOTE: This manual tracking tool yields the positions P(i,t) of cell i at time t in a text file, and invoked as
cm_track.py -i images/ -o track.dat
where the time-lapse images are in the folder images/ and the position data is collected in the file track.dat (Figure 4).
- Perform cell tracking with a custom-developed python tool (http://github.com/donnagreta/cm_track), which also provides basic editor functions such as deleting trajectory segments.
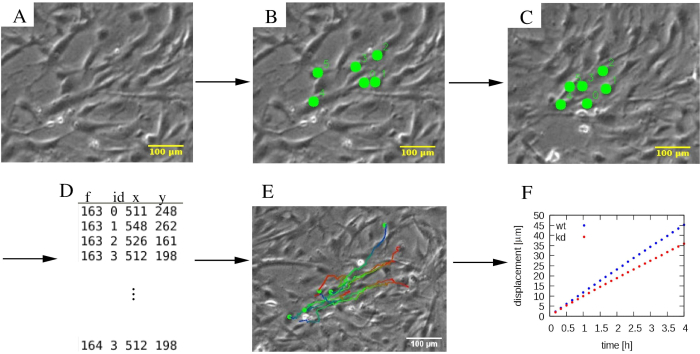
Figure 4: Analysis of individual cell trajectories. (A) Phase-contrast time-lapse micrographs are subjected to (B, C) a manual tracking procedure, which marks cells (green dots). (D) Cell positions (x,y) are stored for each cell distinguished by its ID and for each frame f. (E) Trajectories can be overlaid on the micrographs and color-coded to indicate temporal information. As an example, in each trajectory, a blue to red color palette indicates progressively later trajectory segments, with red and blue marking the initial and final cell locations, respectively. (F) Various statistical properties of trajectories, such as the mean square displacement, can be extracted and used to characterize the motility of various cell populations, which in this example include wildtype (wt, blue), and knockdown (kd, red) MEPM cells. The scalebars represent 100 µm. Please click here to view a larger version of this figure.
- Overlay trajectories on images using a second tool, invoked as
visdat.py -d track.dat -i images/ -o overlay/ -l999 -r3 -C2 - Collect the images with the trajectories overlaid in the folder overlay.
NOTE: In this example, cell position data are stored in the file track.dat, while the time-lapse image sequence is within the folder images. The rest of the parameters control the maximal length of the trajectories drawn (-l), the size of the symbols (-r), and the color scheme (-C) used.
- Analysis of individual cell trajectories
- Characterize trajectories by the total path length
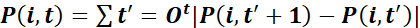 ,
,
and net displacement toward the wound
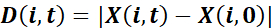 ,
,
where X denotes the projection of P in the direction perpendicular to the wound: the x coordinate of the positions when the wound is parallel to the y axis. - Calculate the guidance efficiency as
 for each cell i and time point t37. Characterize cultures by the population average of these single cell measures, evaluated at a suitable time point t.
for each cell i and time point t37. Characterize cultures by the population average of these single cell measures, evaluated at a suitable time point t.
- Characterize trajectories by the total path length
- Collective streaming motion of cells
- Characterize the local spatial correlations of cell movements by the average velocity of other cells that are in the vicinity of a moving cell, as described previously38,39.
NOTE: The computational code is available at https://github.com/aczirok/flowfield.- Align a reference system with directions front, rear, left, and right to each vector v(x,t) (Figure 5A). Assign each of the surrounding cell or PIV velocity vector to the appropriate spatial cell of the reference system (Figure 5B). Repeat the procedure as each vector serves as the origin of the reference systems (Figure 5C,D) so that a given velocity vector is assigned to multiple bins.
NOTE: A data point could be in front of a vector, and to the left of another one. - Rotate the reference systems into a common orientation (Figure 5C,F) and pool them (Figure 5G).
NOTE: The average of each bin of the pooled velocity data is a velocity vector (U) that is indicative of spatial correlation: the average is a measure of a shared velocity component (Figure 5H). - Fit the U(x) flow fields with an exponential function
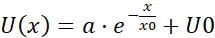 , where a, x0 and U0 are fitting parameters. Out of the three fitting parameters, focus on x0, the correlation length (Figure 5I,J), which is the characteristic distance where local velocity-velocity correlations disappear.
, where a, x0 and U0 are fitting parameters. Out of the three fitting parameters, focus on x0, the correlation length (Figure 5I,J), which is the characteristic distance where local velocity-velocity correlations disappear.
- Align a reference system with directions front, rear, left, and right to each vector v(x,t) (Figure 5A). Assign each of the surrounding cell or PIV velocity vector to the appropriate spatial cell of the reference system (Figure 5B). Repeat the procedure as each vector serves as the origin of the reference systems (Figure 5C,D) so that a given velocity vector is assigned to multiple bins.
- Characterize the local spatial correlations of cell movements by the average velocity of other cells that are in the vicinity of a moving cell, as described previously38,39.
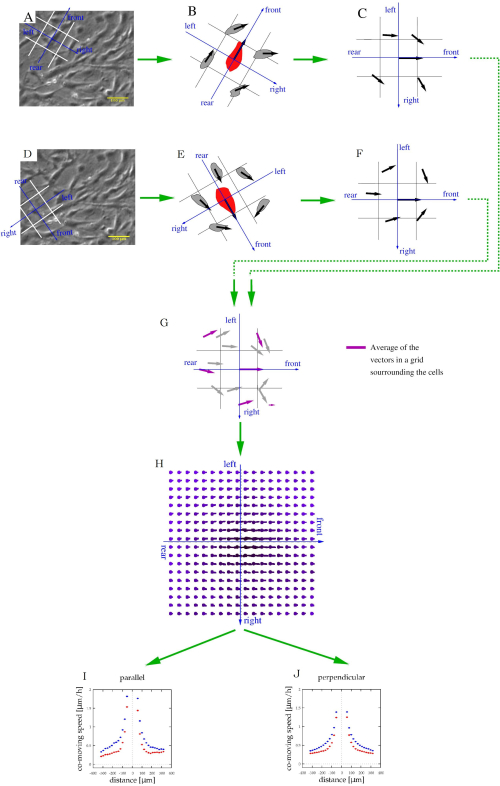
Figure 5: Characterization of stream formation of cultured cells. (A,D) Phase-contrast time-lapse images from Figure 4A are used to identify cell movements. For each moving cell, a frame of reference (blue) and spatial bins (white) were co-aligned to categorize adjacent cells as being in the front, rear, left, or right. (B,E) The velocity of adjacent cells (black vectors) was related to the same frame of reference (C,F). This procedure was repeated for each cell and time-point. (G) After pooling this local information, each bin will contain multiple velocity vectors (gray), which can be averaged to determine the average co-moving velocity (magenta arrows) at various locations relative to an average motile cell. (H) The average velocity map thus characterizes the typical cell velocities at various locations relative to a moving cell. (I,J) Finally, this field was sampled along the front-rear (parallel) axis and also along the left-right (perpendicular) axis. Please click here to view a larger version of this figure.
The dissection of palatal shelves is illustrated in Figure 1. The sequence of incisions is designed to minimize slippage of the tissue. Following the removal of the head (Figure 1A,B), the lower jaw is removed (Figure 1B,C). The incision of the upper part of the head (Figure 1C,D) is done to stabilize the tissue when placed upside down (Figure 1E) to visualize (Figure 1E, dotted lines), pinch (Figure 1F), and excise (Figure 1G) the palatal shelves.
The excised palatal shelf pair from a single embryo is trypsinized and cultured in a 35 mm dish or in a well of a 6-well dish. Larger dishes are not preferred as the cell density is too low for optimal growth. Upon confluence, the cells from each well are trypsinized and passaged into three 35-mm-equivalent wells (passage #1). Confluent cells from passage #1 can then be frozen down into aliquots of 1 × 106 cells/mL. Frozen aliquots are subsequently brought up in a 35-mm-equivalent dish and grown to confluence. The cells are then trypsinized (passage #2) and seeded according to the experiment. Creating and using frozen aliquots helped to normalize conditions, especially with respect to cell density. The use of fresh MEPM cells resulted in more variability in final cell density in experiments, which is believed to be due to a variable proportion of viable or sub-viable cells in fresh cultures. In addition, the number of MEPM cell passages were strictly limited to two (listed above) for these experiments.
Considering that a) cell density was critical, b) MEPM cells from a single embryo were limited, and c) imaging optics were better in a large dish, 2-well silicone inserts were used (Figure 2 and Figure 3). For wound-repair assays, MEPM cells were grown in the 2-well silicone inserts until high confluence, then the inserts were removed, and the wound imaged until closure (Figure 3). However, for 2D culture, the MEPM cells needed imaging as they grew, so the 2-well silicone inserts were simply trimmed to 1/3rd of their height to allow for clear imaging (Figure 2C). Small 3D printed rings40 in 35 mm dishes were also used for 2D motility analysis (Figure 2D).
MEPM trajectories (Figure 4E) are persistent: the direction of cell motility is maintained for several hours. The mean displacement vs. time analysis (Figure 4F) indicates the persistence in the form of displacement being proportional with elapsed time. The typical speed of MEPM cells on tissue culture plastic surface, 10 µm/h, can be also used for quality control of the cell culture. Flow analysis of the motility data reveals that the co-moving clusters of MEPM cells are ~300 µm in size (Figure 5I,J). The representative results also indicate a profound motility difference between wild-type and mutant MEPM cells. In addition to wildtype and mutant comparison, both 2D and wound-repair assays can be combined with various biochemical treatments. For example, PI3K-AKT pathway activators have been used with MEPM cells, as described previously 21.
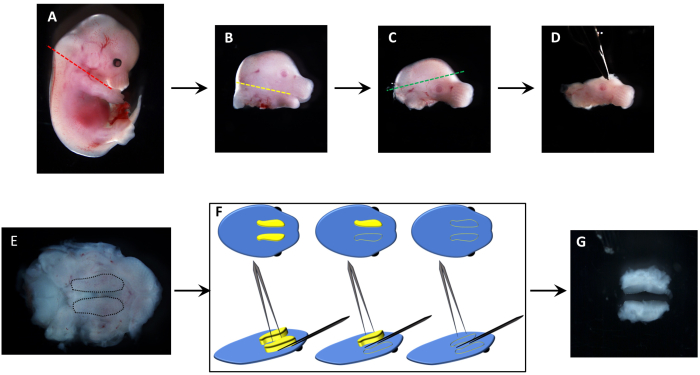
Figure 1: Dissection of embryonic palatal shelves to isolate mesenchymal cells. (A) An E13.5 mouse embryo head is removed by cutting along the neck (red line). (B) Next, the lower jaw and tongue are removed with incisions along the oral cavity, between the upper and lower jaws (yellow line). (C) To stabilize the tissue for palatal shelf removal, the top of the head is excised (green line). (D) The resulting dissected upper jaw region is placed (E) upside down to visualize the palatal shelves (black dotted lines). (F) The excision of individual palatal shelves is depicted schematically, where individual protruding shelves (yellow) are pinched off from the maxilla (blue). (G) Excised pair of palatal shelves from a single embryo, which can then be trypsinized and cultured in a 35 mm dish or in a 6-well plate. Please click here to view a larger version of this figure.
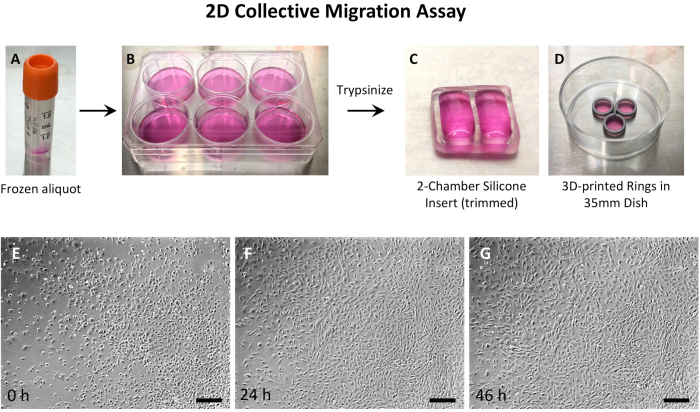
Figure 2: Experimental setup of 2D MEPM cell culture. (A) Thaw a frozen aliquot of MEPM cells, and (B) culture the cells in a 35 mm dish or a 6-well plate. When confluent, trypsinize the cells and seed them as described in the protocol in a 35 mm dish either with (C) 2-well silicone inserts that have been trimmed or (D) with 3D-printed rings. A small culture space minimizes the need for large numbers of MEPM cells. The low profile of the trimmed silicone inserts or the 3D-printed rings allows for direct imaging without a halo effect. Time-lapse imaging can continue until the desired cell density is achieved, which can be up to 72 h. Representative images are shown at (E) 0 h, (F) 24 h, and (G) 45 h timepoints. The images were taken using a 4x objective. The scalebars for E, F, G = 300 µm. Abbreviations: 2D = two-dimensional; MEPM = mouse embryonic palatal mesenchyme; 3D = three-dimensional. Please click here to view a larger version of this figure.
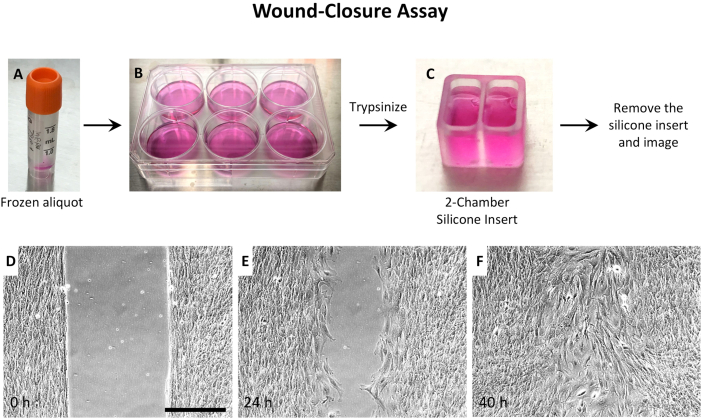
Figure 3: Experimental setup of wound-closure assay using MEPM cells. (A) Thaw a frozen aliquot of MEPM cells, and (B) culture cells in a 35 mm dish or a 6-well plate. When confluent, trypsinize the cells and (C) seed them in the 2-well silicone inserts in a 35 mm dish. Culture the cells in the insert until the desired confluence is achieved (~48 h), then remove the silicone insert and image. Representative images are shown (D) immediately after removal of the insert, (E) after 24 h, and (F) after 40 h. Wound closure takes around 36 h. The images were taken using a 10x objective. The scalebar represents 300 µm. Abbreviations: MEPM = mouse embryonic palatal mesenchyme. Please click here to view a larger version of this figure.
