The ribosome profiling technique (RIBO-seq) was developed in the laboratory of Jonathan Weissman at the University of California, San Francisco1. In comparison to other methods used to study gene expression at the translational level, RIBO-seq focuses on each ribosome binding to mRNA and provides information about its location and the relative number of ribosomes on a transcript. It enables monitoring the process of protein synthesis in vivo and can provide single codon resolution and accuracy allowing the measurement of the ribosome density on both, the individual mRNA and along the entire transcriptome in the cell. At the foundation of the RIBO-seq technique lies the fact that during translation the ribosome binds the mRNA molecule and thus protects the buried fragment of the transcript from a ribonuclease digestion. Upon addition of the ribonuclease, the unprotected mRNA is digested and the fragments enclosed by ribosomes – typically of ~28-30 nt long – remain intact. These fragments, called ribosomal footprints (RF), can then be isolated, sequenced and mapped onto the transcript they originated from resulting in the detection of the exact position of the ribosomes. In fact, the ribosome ability to protect mRNA fragments has been used since the 1960s to study ribosomal binding and translation initiation sites (TIS)2,3,4. However, with the advancement in deep sequencing technology, RIBO-seq has become a gold standard for translation monitoring5 which, through the ribosome engagement, can provide a genome-wide information on protein synthesis6. Ribosome profiling filled the technological gap that existed between quantifying the transcriptome and the proteome6.
To conduct ribosome profiling we need to obtain cell lysate of the organism that had grown under the investigated conditions. Disrupting these conditions during cell collection and lysis may provide unreliable data. To prevent this, translation inhibitors, rapid harvesting and flash freezing in liquid nitrogen are commonly used. Cells can be lysed by cryogenic grinding in a mechanical homogenizer like a mixer mill7,8 or a bead beater9, and by trituration through a pipette10 or with a needle11. The lysis buffer can be added just before or shortly after pulverization of the cells. In our protocol we use liquid nitrogen to precool mortar and pestle, as well as aluminum oxide as a gentler approach to disruption of the bacterial cell wall, which prevents RNA shearing often encountered when methods such as sonification are applied. After pulverization, we add an ice-cold lysis buffer into the cooled contents of the mortar. Selection of an appropriate lysis buffer is important for obtaining the best resolution of ribosomal footprints. Since ionic strength affects both the RF size and the reading frame precision, it is currently recommended to use lysis buffers with low ionic strength and buffer capacity, even if it appears that buffer composition does not affect ribosomal occupancy on mRNAs11,12. Important components of the lysis buffer are magnesium ions, the presence of which prevents dissociation of the ribosomal subunits and inhibits spontaneous conformational changes in the bacterial ribosomes11,13. Calcium ions also play a significant role and are essential for the activity of micrococcal nuclease (MNase) used in the bacterial ribosome profiling method14. Addition of guanosine 5′-[β,γ-imido]triphosphate (GMP-PNP), a non-hydrolyzable analog of GTP, together with chloramphenicol inhibits translation during lysis15.
When the lysate is obtained, it is clarified by centrifugation and divided into two portions, each for a RIBO-seq and a high-throughput total mRNA sequencing (RNA-seq) since they are performed simultaneously (Figure 1). RNA-seq provides a point of reference which enables the comparison of data from both RIBO-seq and RNA-seq during data analysis. The investigated translatome is defined by normalization of ribosomal footprints to mRNA abundance16. Data from RNA-seq can also help identify cloning or sequencing artifacts17.
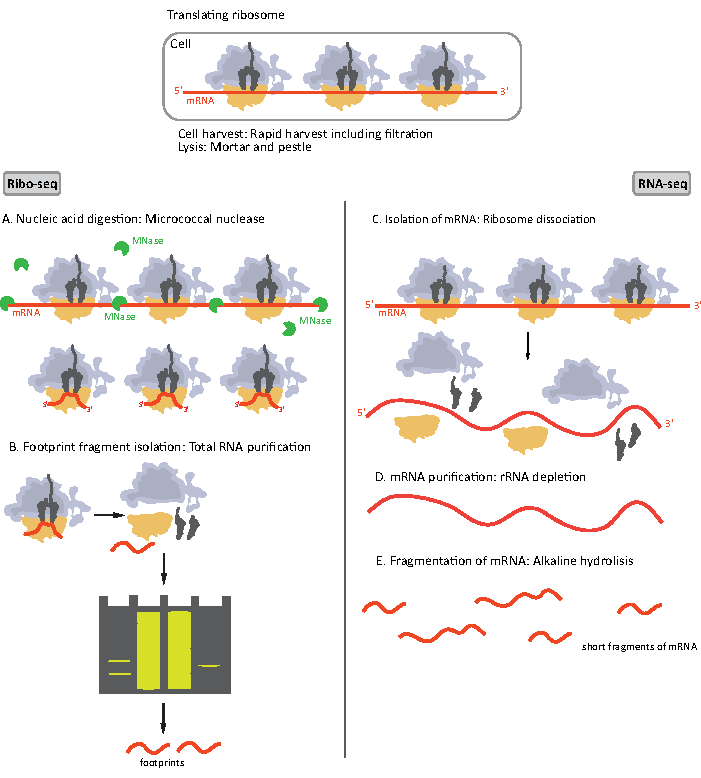
Figure 1. Schematics of mRNA sample preparation for RIBO-seq and RNA-seq. For RIBO-seq library preparation, RNA is digested with MNase (A), followed by the size selection of RF of ~28-30 nt length (B); for RNA-seq RNA is isolated (C), depleted of rRNA (D), and the resulting mRNA is randomly fragmented into fragments of varying lengths (E). Please click here to view a larger version of this figure.
Initial steps of the procedure of sample preparation for RIBO-seq and RNA-seq differ slightly (Figure 1). For the ribosomal profiling, the lysate needs to be digested by a specific endonuclease to degrade the mRNA molecules not protected by the ribosomes. In standard protocols, the obtained monosomes are recovered by a sucrose cushion ultracentrifugation or a sucrose gradient ultracentrifugation8,14. In this article, we show that this step is not necessary to isolate RF required for the RIBO-seq in bacteria, likewise for eukaryotic cells18, and that size selection of the appropriate length mRNA fragments from the polyacrylamide gel is sufficient.
For RNA-seq, mRNA is obtained by the depletion of rRNA from the total RNA – rRNA molecules hybridize to the biotinylated oligonucleotide probes which bind to the streptavidin-coated magnetic beads. The rRNA-oligonucleotide-bead complexes are then removed from the sample with a magnet resulting in a rRNA depleted sample19,20. The purified mRNA molecules are then randomly fragmented by alkaline hydrolysis. The obtained fragments of mRNA as well as the ribosomal footprints are converted into cDNA libraries and prepared for deep sequencing (Figure 2). This involves ends repair needed after alkaline hydrolysis (for mRNA) and enzymatic digestion (for RF): dephosphorylation of 3' ends followed by phosphorylation of 5' ends. The next steps are adaptors ligation and the reverse transcription to create cDNA inserts framed by sequences required for the next generation sequencing (NGS) using Illumina platform. The last phase of library preparation is a PCR reaction in which the constructs are amplified and labelled with sample specific barcodes to allow multiplexing and sequencing various samples on one channel. Before sequencing, the quality and quantity of the libraries are assessed by the high-sensitivity DNA on-chip electrophoresis. cDNA libraries with appropriate parameters can then be pooled and sequenced. Sequencing can be performed on different Illumina platforms, such as MiSeq, NextSeq or HighSeq, depending on the number of libraries, required read length and sequencing depth. After sequencing, the bioinformatic analysis is performed.

Figure 2. Library preparation. Library preparation includes the ends repair, adapters ligation, reverse transcription and amplification with barcoding. Please click here to view a larger version of this figure.
The ribosome profiling is a universal method which can be easily modified and adjusted according to the scientific question. Originally it was used in yeast1, but shortly after it was applied to bacterial cells21 as well as eukaryotic model organisms including mouse10, zebrafish22, fruit fly23 and Arabidopsis thaliana24. It was also used for studying different ribosome types: cytoplasmic, mitochondrial25,26 and chloroplast27,28. In eukaryotes RIBO-seq is commonly adapted and refined to investigate specific aspects of translation, including initiation10,11,29,30,31,32, elongation1,10,11,31,33, ribosome stalling33 and conformation change33. Most of the modifications involve the use of different translation inhibitors. In bacteria however, analogous studies have been difficult to conduct because of the paucity of inhibitors with the required mechanism of action34. The most commonly used translation inhibitor in bacteria is chloramphenicol (CAM) which binds to the peptidyl transferase center (PTC) and prevents correct positioning of the aminoacyl-tRNA in the A-site. As a result, CAM prevents the formation of a peptide bond which leads to arresting the elongating ribosomes35. Other examples of translation inhibitors in bacteria are tetracycline (TET)36, retapamulin (RET)34 and Onc11237 which have been used to investigate translation initiation sites. TET, which prevents tRNA delivery to the ribosome by directly overlapping with the anticodon stem-loop of tRNA at the A-site, was originally applied to verify the results obtained from CAM treatment since they are both antibiotics inhibiting translation elongation38. TET was found to detect primary TIS, however was unable to reveal internal TIS36. RET binds in the PTC of the bacterial ribosome, and prevents formation of the first peptide bond by interfering with an elongator aminoacyl-tRNA in the A site. Applying RET results in ribosomes arrest at both primary as well as internal TISs34. Onc112, a proline-rich antimicrobial peptide, binds in the exit tunnel and blocks aminoacyl-tRNA binding in the ribosomal A site. As a result, Onc112 prevents initiation complexes from entering the elongation phase37.
The main information ribosome profiling provides is ribosomes density and their position on the mRNA. It was successfully applied to investigate differential gene expression at the level of translation in various growth conditions1,6, measure translational efficiency1,38,39 and detect translation regulation events such as ribosomal pausing10. RIBO-seq also allows for uncovering the translation of annotated ncRNA, pseudogenes and unannotated small open reading frames (ORF) leading to the identification of novel and/or very short protein coding genes10,12,22,30,37. In such cases, RIBO-seq can fine-tune and improve genome annotation. With its high sensitivity for the identification of translated ORFs and its quantitative nature, ribosome profiling can also serve as a proxy for the proteome determination or in aiding proteomics studies31,34,39. By mapping TIS, ribosome profiling reveals N-terminally extended and truncated isoforms of known proteins10,32. RIBO-seq was also adapted to study co-translational folding of proteins14,21,24. This method enables measuring of elongation rates1,10,39 or decoding speeds of individual codons6 and helps in developing quantitative models of translation17. The ribosome profiling method is also capable of providing mechanistic insights into the ribosome pausing in bacteria7,15,17, frameshifting40, stop-codon readthrough21, termination/recycling defects41,42 and ribosomal conformation changes33 in eukaryotes. RIBO-seq was also adapted to examine the impact of specific trans-acting factors on translation such as miRNAs6 and RNA-binding proteins in eukaryotes16,43. However, it is important to acknowledge that the experimental design and the obtained resolution of RIBO-seq determine the amount of information that can be extracted from the resulting sequencing data12.
The exemplary results presented here were obtained in a study examining translation regulation in sporulating WT Bacillus subtilis cells. Overnight cultures were diluted to OD600 equal to 0.1 in 100 mL of rich medium and incubated at 37 °C with vigorous shaking until OD600 reached 0.5-0.6. The rich medium was then replaced with minimal medium to induce sporulation process and the incubation was continued for up to four hours. Cells were harvested every hour beginning with T0 – sporulation induction – resulting in five time points. One minute before harvesting, the cultures were treated with chloramphenicol as described in the protocol above. Cells were collected by filtration in a prewarmed glass filtration system using 0.45 µm mixed cellulose esters membrane (MCE) filters and flash-frozen in liquid nitrogen.
The amount of ribonucleic acid obtained in the lysate depends on growth conditions, culture volume and density. Following our protocol, 100 mL of the bacterial culture (Bacillus subtilis) yields 1-4 mg of RNA on average. Examples of lysates obtained from the experiment described above are shown in Table 1.
| RNA concentration [ng/µL] | RNA amount [mg] | A260/A280 | A260/A230 | |
| WT0 | 5845 | 3.21 | 1.57 | 0.83 |
| WT1 | 6275 | 3.45 | 1.58 | 0.79 |
| WT2 | 3525 | 1.94 | 1.38 | 0.56 |
| WT3 | 3717 | 2.04 | 1.37 | 0.61 |
| WT4 | 1653 | 0.91 | 1.11 | 0.43 |
Table 1. RNA concentration and amount in representative samples of B. subtilis lysates. The table includes samples from the wild type (WT) B. subtilis which were harvested before the sporulation induction (0) and one hour (1), two (2), three (3) or four (4) hours post sporulation induction. Lysis was performed with 550 µL of lysis buffer containing chloramphenicol. It can be noticed that as the sporulation proceeds the amount of obtained RNA decreases.
When the amount of RNA is lower than 1 mg, 0.25 mg of RNA is used for RIBO-seq instead of 1-0.5 mg and the amount of MNase is then adjusted respectively. The representative RNA amount and concentration after nuclease digestion is shown in Table 2. The average amount of RNA after digestion and cleaning is 50 µg from 0.5 mg of RNA input.
| RNA concentration [ng/µL] | RNA amount [µg] | A260/A280 | A260/A230 | |
| WT0-R | 1015 | 50.75 | 2.12 | 1.84 |
| WT1-R | 1157.4 | 57.87 | 2.10 | 2.19 |
| WT2-R | 847.2 | 42.36 | 2.10 | 2.00 |
| WT3-R | 983.6 | 49.18 | 2.09 | 2.16 |
| WT4-R | 206 | 10.30 | 2.13 | 1.00 |
Table 2. The amount and concentration of RNA after nuclease digestion of the representative samples for RIBO-seq. The table includes samples from the wild type (WT) B. subtilis which were harvested before sporulation induction (0) and one hour (1), two (2), three (3) or four (4) hours post sporulation induction; the letter "R" refers to samples for RIBO-seq. Because RNA concentration of WT4-R sample was low, 0.25 mg of RNA was digested, while 0.5 mg of the RNA for the remaining samples was used for ribosomal footprint generation. The amount of MNase for digestion was 178.125 U for 0.25 mg of RNA in WT4-R, and 356.25 U for 0.5 mg of RNA in other samples. After digestion the samples were cleaned with the commercial RNA clean-up kit and ribonucleic acid concentration was measured with the NanoDrop.
After MNase digestion, size selection is performed with the aid of PAGE. An example of a 15% TBE-urea polyacrylamide gel with the digested samples is shown in Figure 3. Fragments of gel between 26 and 32 nt were excised with a sterile razor blade and incubated overnight in the overnight incubation buffer. The next day, ribosomal footprints were cleaned with the commercial RNA clean-up kit.

Figure 3. 15% TBE-urea polyacrylamide gel with representative samples after MNase digestion. The figure includes samples from the wild type (WT) B. subtilis which were harvested before sporulation induction (0) and one hour (1), two (2), three (3) or four (4) hours post sporulation induction; the letter "R" refers to samples for RIBO-seq. 15 µL of denatured samples were loaded into the gel wells in order: WT2-R, WT3-R, WT4-R, WT0-R, WT1-R and the remaining volume was stored at -80 °C as a back-up. 29 nt oligonucleotide and mix of 26 nt and 32 nt oligonucleotides were used as markers. Electrophoresis was performed as described above and the gel was stained in a SYBR Gold-bath. Please click here to view a larger version of this figure.
Parallel to nuclease digestion, the lysates for RNA-seq were cleaned with the commercial RNA clean-up kit and the obtained concentrations of RNA were measured with NanoDrop. The representative results are presented in Table 3. After cleaning, the amount of ribonucleic acid decreased to 40-500 µg.
| RNA concentration [ng/µL] | RNA amount [µg] | A260/A280 | A260/A230 | |
| WT0-m | 10274.1 | 513.70 | 2.14 | 2.35 |
| WT1-m | 10118.3 | 505.92 | 2.12 | 2.33 |
| WT2-m | 5599.7 | 279.98 | 2.15 | 2.29 |
| WT3-m | 6562.2 | 328.11 | 2.08 | 2.31 |
| WT4-m | 757 | 37.85 | 2.01 | 1.95 |
Table 3. RNA amount and concentration of samples for RNA-seq after RNA purification. The table includes samples from the wild type (WT) B. subtilis which were harvested before sporulation induction (0) and one hour (1), two (2), three (3) or four (4) hours post sporulation induction; the letter "m" refers to samples for RNA-seq (mRNA).
The obtained RNA concentrations of the samples for RNA-seq were used to define RNA input for rRNA depletion. 8 µg of RNA from each RNA-seq sample were used with MICROBExpress bacterial mRNA purification kit (see Table of Materials) according to the manufacturer's protocol and were cleaned with the commercial RNA clean-up kit afterwards. The representative results of rRNA depletion are shown in Table 4.
| RNA concentration [ng/µL] | A260/A280 | A260/A230 | RNA amount [µg] | |
| WT0-m | 62.7 | 1.99 | 1.75 | 3.1 |
| WT1-m | 59.3 | 2.00 | 2.16 | 2.9 |
| WT2-m | 93.7 | 1.97 | 1.69 | 4.7 |
| WT3-m | 68.3 | 1.99 | 2.33 | 3.4 |
| WT4-m | 85.0 | 1.97 | 2.00 | 4.2 |
Table 4. The representative RNA amount and concentration after rRNA depletion. The table includes samples from the wild type (WT) B. subtilis which were harvested before sporulation induction (0) and one hour (1), two (2), three (3) or four (4) hours post sporulation induction; the letter "m" refers to samples for RNA-seq. 8 µg of RNA from samples for RNA-seq were used for rRNA depletion and cleaned with a commercial RNA clean-up kit afterwards.
Next, the obtained mRNA was fragmented by alkaline hydrolysis and cleaned with a commercial kit as described in the protocol. The fragmented mRNA and ribosomal footprints were dephosphorylated at 3' ends and phosphorylated at 5' ends according to the above protocol. The samples were then used to construct cDNA libraries for Illumina sequencing, as described in the protocol. Figure 4 shows an example of a 6% polyacrylamide gel with the obtained libraries. Gel fragments in the range of 135-180 nt for RNA-seq and 135-170 nt for RIBO-seq were excised with sterile razor blades and incubated overnight in nuclease-free water. Libraries were cleaned with a commercial DNA clean-up kit and quality and quantity was assessed by a high-sensitivity DNA on-chip electrophoresis using the Agilent 2100 Bioanalyzer.

Figure 4. 6% polyacrylamide gel of cDNA libraries before and after size selection. Quick-Load pBR322 DNA-MspI Digest was used as a ladder. 25 µL of samples were loaded into the gel in the following order: three RIBO-seq libraries and six RNA-seq libraries. The remaining volumes of samples were stored at -80 °C as a back-up. The figure includes samples from the wild type (WT) B. subtilis which were harvested one hour (1), two (2), three (3) or four (4) hours post sporulation induction; the letter "R" refers to samples for RIBO-seq and the letter "m" refers to samples for RNA-seq. Please click here to view a larger version of this figure.
The examples of virtual gel images and electropherograms obtained from the Agilent 2100 Bioanalyzer are shown in Figure 5. Bands and peaks representing the RIBO-seq libraries are more narrow and better defined compared to these representing RNA-seq libraries. According to the on-chip electrophoresis, libraries are approximately 150 bp long, which is an expected library size. The adapters and the barcode used in library preparation are 119 nt long and the inserts are either 29-30 nt long (for RIBO-seq) or in a range between 25 and 50 nt (for RNA-seq). The additional peaks closer to the 200 bp obtained from RIBO-seq libraries may indicate PCR artefacts which can be discarded during bioinformatic analysis when the data obtained from sequencing is trimmed and rRNA/tRNA is filtered. The average quantity of DNA is 32 ng providing sufficient quantity of material required for the next generation sequencing.

Figure 5. The examples of electropherograms and virtual gel images from a high-sensitivity DNA on-chip electrophoresis. The figure includes samples from the wild type (WT) B. subtilis which were harvested before sporulation induction (0) and one hour (1), two (2), three (3) or four (4) hours post sporulation induction; the letter "R" refers to RIBO-seq libraries and the letter "m" refers to RNA-seq libraries. Please click here to view a larger version of this figure.
After quality control by the on-chip electrophoresis, libraries were pooled for single-end 50 bp sequencing on the Illumina's NextSeq500 platform (10 libraries per channel, with a minimum of 350 mln reads per channel). Sequencing yielded 30-57 million reads per sample for RNA-seq libraries and 30-50 million reads per sample for RIBO-seq libraries. Trimming the adaptors and poor quality sequences resulted in 24-47 mln reads per sample for RNA-seq samples and 25-50 mln reads per sample for RIBO-seq samples. Mapping, performed with STAR44, yielded 2.4-9.6 million of uniquely mapped reads per sample for RNA-seq samples (which constitute 8-16.8% of the total number of reads) and 2.3-10.4 million uniquely mapped reads (7.7-22.1%) for RIBO-seq samples. The obtained data was quality control checked with FastQC45. Examples of the quality plots produced by FastQC for the trimmed sequences are shown in Figure 6. The sequences of the length of interest, which is <32 nt for RIBO-seq and <50 nt for RNA-seq, presented very good quality. The examples of plots of GC content and distributions of sequence lengths over all trimmed sequences are shown in Figure 7. The additional peak at 72% in GC distribution plot of RIBO-seq library may potentially indicate rRNA contamination which can be removed during bioinformatic analysis by rRNA filtration46,47,48. The sequence lengths distribution is very narrow for RIBO-seq libraries, with a peak at 26-27 nt whereas the length distribution for RNA-seq libraries is broader and ranges between 15 and 50 nt.

Figure 6. The examples of box-and-whisker plots of quality scores across all bases for RIBO-seq and RNA-seq libraries after trimming. The figure includes samples from the wild type (WT) B. subtilis which were harvested before sporulation induction (0) and four (4) hours post sporulation induction; the letter "R" refers to RIBO-seq samples and the letter "m" refers to RNA-seq samples. Please click here to view a larger version of this figure.

Figure 7. The examples of plots of GC content and sequence length distribution of all sequences for RIBO-seq and RNA-seq libraries after trimming. The figure shows samples from the wild type (WT) B. subtilis which were harvested four (4) hours post sporulation induction; the letter "R" refers to sample for RIBO-seq and the letter "m" refers to sample for RNA-seq. Please click here to view a larger version of this figure.
To verify whether the RIBO-seq data provides information on true ribosomal footprints rather than random mRNA fragments of the selected length, we compared sequencing data from RIBO-seq and RNA-seq using RiboToolkit, a web server for RIBO-seq data analysis49. Ribo-seq data exhibits triplet periodicity and a tall, narrow peak corresponding to the initiating ribosomes and characteristic of the RF as shown in Figure 8A, which is not observed in the RNA-seq data (Figure 8B). Moreover, the metagene plot showing the profiles of average coverage of RF and mRNA fragments on the coding sequences (CDSs) revealed a higher proportion of reads mapped to the CDSs in the RIBO-seq dataset (Figure 8C), as expected.

Figure 8. Metagene periodicity plots and average coverage of RF and mRNA fragments on CDSs. Metagene profiles were obtained with RiboToolkit, an integrated web server49. CDS length was normalized to a value of 1 and each CDS was divided into 100 equal bins. The figure shows samples from the wild type (WT) B. subtilis which were harvested one (1) hour post sporulation induction; the letter "R" refers to sample for RIBO-seq and the letter "m" refers to sample for RNA-seq. Please click here to view a larger version of this figure.
To compare whether the RIBO-seq protocol presented here yields results similar to those obtained by standard method of RF recovery (sucrose gradient ultracentrifugation), we paralleled the sequencing results from libraries prepared following the two methods. The experiments investigating translation regulation during sporulation in the WT B. subtilis were conducted according to the presented RIBO-seq protocol, as well as following the standard protocol which includes monosomes recovery by a sucrose gradient ultracentrifugation. The results of ribosome coverage profiles obtained from the two experiments are shown in Figure 9. The visualisation of mapped RFs is very similar between the two experiments and yielded similar results upon bioinformatic analysis.

Figure 9. The visualisation of obtained RF mapped onto the genome. The figure includes RF obtained from wild type B. subtilis harvested four hours post sporulation induction and prepared according to the standard protocol (WT4-R-s, upper panel) or the presented protocol (WT4-R, lower panel). Please click here to view a larger version of this figure.
