The most common application of bead loading is to introduce one or more types of protein into adherent human cells. To illustrate this, cells were bead-loaded with a solution of a Cy3- and Alexa488-conjugated Fab protein. Although not every cell in the microwell was bead-loaded, the cells that were loaded almost always had both Cy3- and Alexa488-labeled proteins together (Figure 2A). According to an earlier estimate, when 0.5 microgram of Fab diluted in 4 microliters is bead-loaded29, as in Figure 2A, each cell is loaded with roughly 106 Fab molecules.
Plasmid DNA encoding GFP (1 µg of plasmid DNA, 1.8 µL of a 557 ng/µL solution) and 0.5 µg of Cy3-labeled Fab was also introduced into cells via bead loading and subsequently expressed and visualized (Figure 2B). The GFP fluorescence indicated that the GFP-encoding plasmid was not only loaded into cells but also expressed. Thus, in the same cell, bead loading can introduce a protein probe (e.g., Cy3-labeled Fab) and reporter plasmid (e.g., GFP), as performed in this laboratory previously22,23,24. We determined that 40% of the cells were bead-loaded with Fab protein and 21% of the bead-loaded cells expressed the co-loaded plasmid, as shown in the representative fields-of-view in Figure 2B. Typically, each chamber is loaded with 1-2 µg of plasmid, approximately the same amount as lipofection.
Bead-loaded cells express widely varying levels of plasmids (Figure 2C,D). To specifically measure this, we used the Fisher Ratio test to compare the distributions of protein and plasmid intensity data. The results showed that although proteins 1 and 2 had similar intensity distributions (p = ~1), each protein had a significantly smaller distribution than the plasmid (p = 3.2e-6 and 1.8e-5). Although this could be due to variability in how many plasmids are loaded per cell, the greater source of variability may arise from the many steps required for plasmid expression that are likely to vary greatly between cells, including being imported into the cell nucleus, transcription, and translation. In contrast, the levels of bead-loaded proteins had slight cell-to-cell variance, and the levels of two simultaneously loaded proteins were highly correlated with each other (Figure 2D,E).
Plasmid expression can be seen as early as 2-4 h post bead loading but may occur later depending on when optimal plasmid expression is obtained. We recommend performing a time course to determine the best window of expression for a specific plasmid spanning 2-24 h post bead loading. This can be done in one chamber with long timeframe imaging or by bead loading and staggering multiple chambers. Bead-loaded cells remain adherent and are healthy enough to grow and divide. Bead-loaded human U2OS cells were imaged directly before, directly after, and 24 h after bead loading. Proper bead loading had almost no noticeable effect on the number of cells or their morphology, as shown in Figure 3A (left, middle).
In contrast, poor bead loading with too many beads and excessive tapping force is depicted in Figure 3B. This caused much cell loss (large patches of the coverglass without cells and detached, floating, out-of-focus cells), poor cell morphology (cells appearing rounded up and poorly adhered), and clusters of beads remaining on the coverglass after bead loading. Though cells are thought to undergo mechanical damage during bead loading, cells grew and proliferated in the properly bead-loaded chamber, as evidenced by the increased number of cells 24 h after bead loading (Figure 3A, right). The effect on cell viability can be assessed through a variety of assays, such as a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, to compare bead-loaded to mock-loaded cells30. Further, this and previous work show that the bead-loaded cells undergo cell division (Figure 3C and Supplemental Video 1), and the timing of mitosis is not affected by bead loading31, which serves as further evidence for sustained cell health after bead loading.
Bead loading is a versatile technique, accommodating several adherent cell lines and various macromolecules. Here, this variety has been demonstrated by loading RPE1 and HeLa cell lines with Fab (Figure 4A,B). Table 1 provides further examples of bead loading in different cell lines, in this laboratory and beyond, and points out some of the nuanced differences between bead loading protocols from other labs. Of note, the diameter of glass beads used for loading varies greatly between laboratories, though the most efficient loading was found for small, 75 µm diameter beads in several cell lines14. Further, this laboratory has begun bead loading RNA as well (data not shown). Figure 4C displays a representative U2OS cell bead-loaded with a Cy5-RNA 9mer and Cy3-DNA 28mer together.
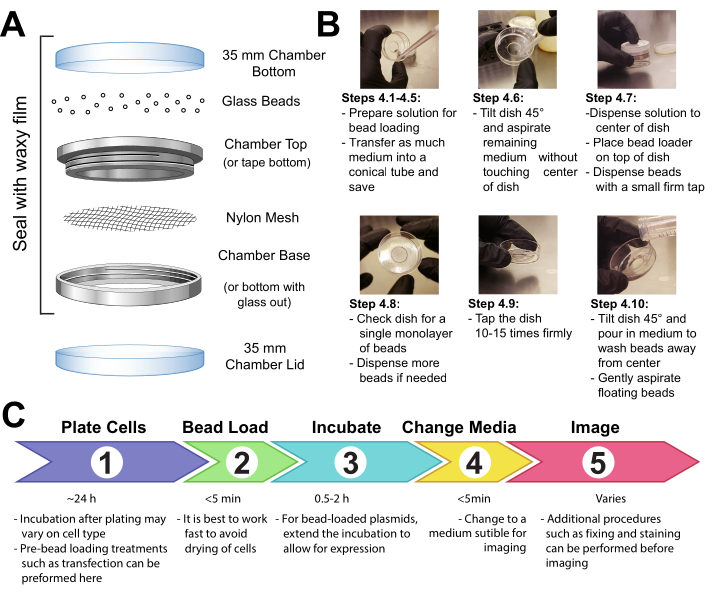
Figure 1: Bead loading apparatus, technique, and timeline Please click here to view a larger version of this figure.
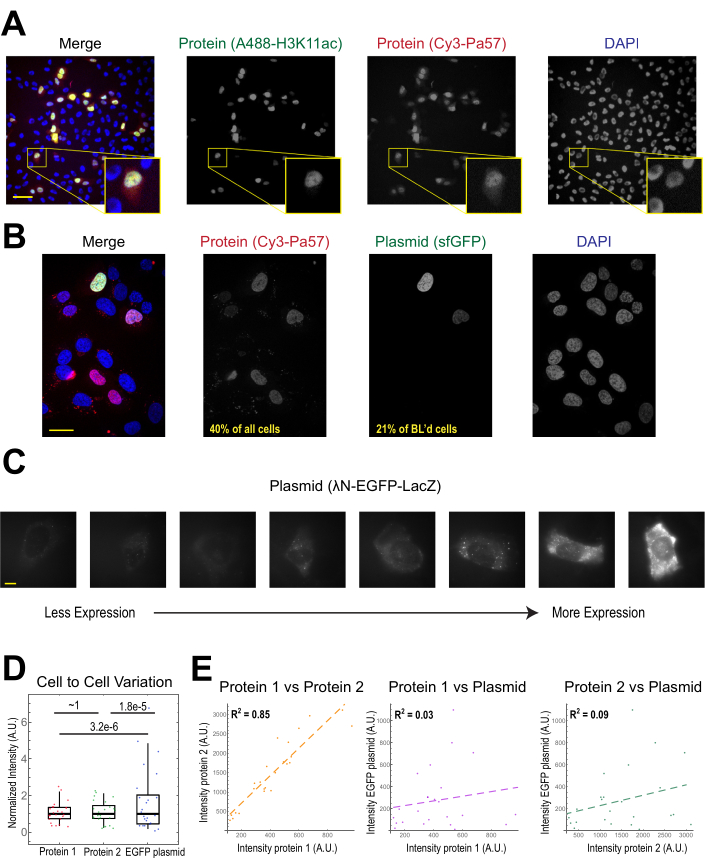
Figure 2. Bead loading introduces low variability in protein concentration but high variability in plasmid expression. (A) Cells were bead-loaded with 0.5 µg of each of Alexa488-conjugated anti-H3K27 acetyl Fab (green) and Cy3-conjugated anti-RNAPII-Serine 5-phosphorylated Fab (red) in 4 µL of bead loading solution. Cells were DAPI-stained (blue) and then live-imaged immediately. Scale bars = 20 µm. (B) Cells were bead-loaded with 0.5 µg of Fab protein (Cy3-conjugated anti-RNAPII-Serine 5-phosphorylated protein, red) and 1 µg of plasmid encoding superfolder GFP-H2B (green) in 4 µL of bead loading solution. After 24 h, cells were DAPI-stained (blue) and imaged live. Scale bars = 30 µm. (C–E) Protein 1 (JF646-HaloLigand-labeled HaloTag-MCP), protein 2 (Cy3-conjugated anti-FLAG Fab), and a plasmid encoding EGFP (λN-EGFP-LacZ) were bead-loaded together into cells. The total intensity in each fluorescent channel was measured in a 1.3 x 1.3 µm patch in the cytoplasm of each cell. N = 25 cells. (C) Representative cells expressing the bead-loaded plasmid, λN-EGFP-LacZ. The same imaging conditions and intensities were used for all cells. Spots are aggregates of the expressed protein. Scale bars = 10 µm. (D) The chart shows each cell's total intensity of either protein 1, protein 2, or EGFP expressed from the plasmid. Each channel was normalized to the median. Bonferroni-corrected P values were calculated by the Fisher Ratio test to determine whether the distribution of protein or plasmid intensity data has the same variability. Each point represents a cell. (E) The total intensities for either both proteins, protein 1 and the plasmid, or protein 2 and the plasmid, are plotted against each other. Calculated R2 values are displayed. Each point represents a cell. Abbreviations: DAPI = 4′,6-diamidino-2-phenylindole; EGFP = enhanced green fluorescent protein; A.U. = arbitrary units; MCP = MS2 coat protein; RNAPII = RNA polymerase II. Please click here to view a larger version of this figure.
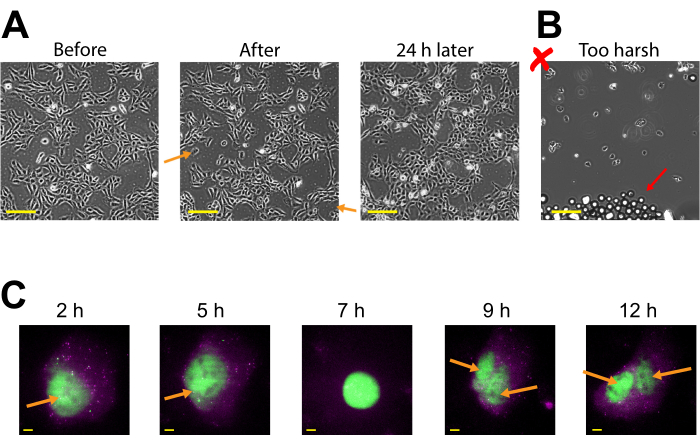
Figure 3: Bead-loaded cells remain adherent and are healthy enough to grow and divide. (A) U2OS cells were bead-loaded with 0.5 µg of Cy3-conjugated anti-FLAG Fab in 4 µL of bead loading solution. The cells were imaged directly before, directly after bead loading, and 24 h after bead loading. Orange arrows identify areas where cells peeled off during bead loading. Scale bars = 2 mm. (B) Representative image of U2OS cells bead-loaded with components from (A) but with harsh tapping and too many beads. The red arrow identifies extra glass beads. Scale bar = 2 mm. (C) U2OS cells were loaded with 1.5 µg of the 14.4 kbp plasmid smFLAG-KDM5B-15xBoxB-24xMS2, 0.5 µg of Cy3-conjugated anti-FLAG Fab (green), 130 ng of HaloTag-MCP (magenta) in 8 µL of bead loading solution. Directly before imaging, the HaloTag was stained with JF646-HaloLigand. The MS2 stem-loops of the mRNA transcribed from the reporter plasmid are labeled by MCP (magenta spots), and FLAG-tagged translated reporter protein is labeled by anti-FLAG Fab (green colocalization to mRNA). Mature Fab-labeled protein localizes to the nucleus. This cell was imaged 4-15 h after bead loading. Yellow arrows identify the cell nucleus before and nuclei after cell division. Scale bars = 5 µm. Abbreviation: MCP = MS2 coat protein. Please click here to view a larger version of this figure.
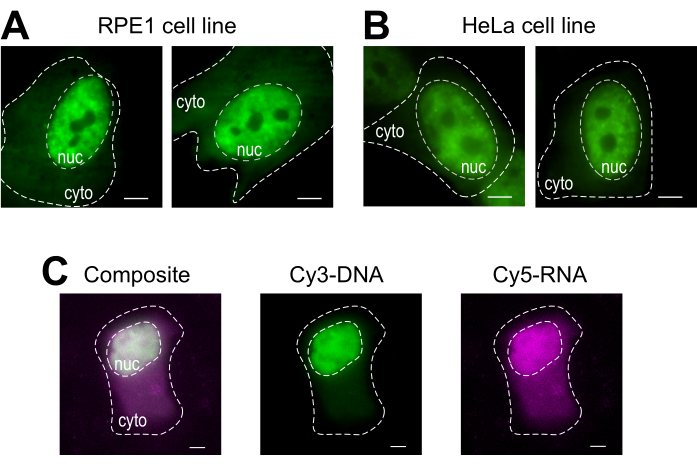
Figure 4: Variations in cell type loading material of the bead loading protocol. (A–B) RPE1 (A) and HeLa (B) cells were bead-loaded with 1.5 µg of a nuclear Fab protein (anti-RNAPII-Serine 5-phosphorylation) in 4 µL of loading solution. Each cell's nucleus (nuc) and cytoplasm (cyto) are marked. Cells were imaged 6 h after being bead-loaded. Scale bars = 5 µm. (C) Human U2OS cells were bead-loaded with both Cy5-RNA 9mer (magenta) and Cy3-DNA 28mer (green) oligos, 10 picomoles of each, in 4 µL of bead loading solution. Cells were imaged 4 h after being bead-loaded. All cell nuclei are highlighted by a dashed line. Scale bars = 5 µm. Abbreviations: RNAPII = RNA polymerase II. Please click here to view a larger version of this figure.
| Cell line | Cell type | Bead loading effectiveness | Notes/reference |
| Stem cells (human) | Embryonic stem cells | Difficult | *Many cells peel off during bead loading if plated on gelatin-coated plates |
| HEK 293 | Human embryonic kidney cells | Difficult | *Need to lay down a gel matrix to imaging chamber surface before bead loading. Tap gently when bead loading at first. |
| Neurons (rat) | Primary embryonic neurons (e-18), dissociated | Very inefficient | *Efficient bead loading of neurons was not observed using this standard bead loading protocol. This could be due to the non-adherent nature of neurons or from consequent damage to neural processes. |
| MDCK (canine) | Madin-Darby canine kidney cells | See McNeil and Warder (1987)14 | *Low-efficiency bead loading14 |
| U2OS (human) | Osteosarcoma | Standard bead loading protocol | |
| HeLa (human) | Cervical cancer | Standard bead loading protocol | |
| RPE1 (human) | Epithelial cells immortalized with hTERT | Standard bead loading protocol | |
| HFF (human) | Primary foreskin fibroblasts | See Besteiro et al. (2009)31 | *Modified protocol of tilting instead of tapping31 |
| BALB/c 3T3, NIH 3T3, and Swiss 3T3 (mouse) | Embryonic fibroblasts | See Gilmore and Romer (1996)32, Emerson et al. (2014)33 and McNeil and Warder (1987)14 | *425–600 μm glass beads reported32 |
| *Used 200–300 μm glass beads33 | |||
| *75 μm glass beads gave better results than 400 μm14 | |||
| DM (Indian muntjac) | Skin fibroblasts | See Manders, Kimura, and Cook (1999)34 | |
| CHO (hamster) | Epithelial-like ovary cells | See Memedula and Belmont (2003)35 | *Used 425–600 μm glass beads35 |
| BAE (bovine) | Bovine aortic endothelial cells (BAEC-11) | See McNeil and Warder (1987)14 | *75 μm glass beads gave better results than 400 μm14 |
| PtK-2 (Potomus tridaclylis) | Epithelial kidney cells | See McNeil and Warder (1987)14 | *75 μm glass beads gave better results than 400 μm14 |
| HUVEC (human) | Umbilical vein endothelial cells | See Gilmore and Romer (1996)32 | *Used 425–600 μm glass beads32 |
| J774 and J774.2 (mouse) | monocyte macrophage cells | See Becker et al. (2005)36 and McNeil and Warder (1987)14 | *Gentle agitation (instead of tapping) and 425–600 μm glass beads36 |
| MS-5 (mouse) | bone marrow stromal cells | See Molenaar et al. (2003)37 | |
| WPE1-NB11 (human) | prostate epithelial cells | See Gilmore and Romer (1996)32 and | *Used 425–600 μm glass beads32 |
| Emerson et al. (2014)33 | *Used 200–300 μm glass beads33 |
Table 1: Bead loading in different cell lines. For the cell lines that have not yet been bead-loaded in this laboratory, references and notes on variations in the protocol are provided.
Supplemental Video 1: Example of a bead-loaded cell undergoing cell division. U2OS cells were loaded with 1.5 µg of the 14.4 kbp plasmid smFLAG-KDM5B-15xBoxB-24xMS2, 0.5 µg of Cy3-conjugated anti-FLAG Fab (green), 130 ng of HaloTag-MCP (magenta) in 8 µL of bead loading solution. Directly before imaging, the HaloTag was stained with JF646-HaloLigand. The MS2 stem-loops of the mRNA transcribed from the reporter plasmid are labeled by MCP (magenta spots), and FLAG-tagged translated reporter protein is labeled via anti-FLAG Fab (green colocalization to mRNA). Mature Fab-labeled protein localizes to the nucleus. This cell was imaged 4-15 h after bead loading. Scale bar = 10 µm. Abbreviation: MCP = MS2 coat protein. Please click here to download this Video.
