NOTE: Use high-performance liquid chromatography (HPLC) grade or MS grade reagents whenever possible to minimize the contaminants that may interfere with MS analysis. Do not touch the CE-MS interface with bare hands during the measurement to avoid the possibility of an electrical shock caused by either the electrophoretic voltage or electrospray voltage.
1. Material preparation
- Modification of fused silica capillary for CE
- Prepare a 5% (w/w) hydroxypropyl cellulose (HPC) solution by dissolving HPC powder (molecular weight [MW]: 100 kDa) in water with continuous stirring at room temperature on a magnetic stirrer for ~12 h or until complete disappearance of solid particles12. Remove any visible air bubbles with an ultrasonicator.
- Mount a fused silica glass capillary (inner diameter [ID]: 50 µm, outer diameter [OD]: 360 µm) of approximately 85 cm length into a CE instrument. Rinse the capillary by continuously infusing an organic solvent, such as acetone13, using the autosampler of CE at an infusion pressure of 40 psi for 10-15 min.
- Fill the cleaned capillary with HPC solution using the autosampler at an infusion pressure of 40 psi (which often takes ~40 min). Infuse air into the HPC-filled capillary at 40 psi to ensure free airflow in the capillary, indicated by the air bubbles ejected from the capillary upon immersion in water.
- Bake the HPC-coated capillary in a temperature-programmable oven (ideally the temperature-controlled column oven of a temperature-programmed gas chromatograph) with nitrogen gas (25 psi) flowing through the capillary, following the temperature program shown in Figure 1.
- Cool the oven to room temperature before taking the capillary out. Use this HPC-modified capillary for CE separation.
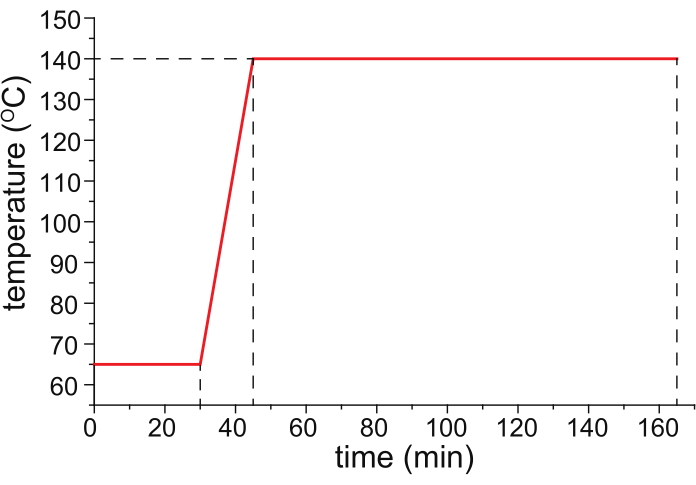
Figure 1: A recommended temperature program for capillary baking. Please click here to view a larger version of this figure.
- Background electrolyte (BGE) solution and modifier solution8
- Prepare 1-10 mL of BGE at the desired concentration (e.g., 10 mM) by dissolving the appropriate quantity of ammonium acetate in 2H2O. Place 200 µL-aliquots of BGE in separate BGE vials and seal the vials with parafilm to minimize the HDX reaction between BGE and water vapor in the air.
- Prepare 10 mL of a modifier solution with 75% (v/v) methanol and 25% (v/v) water, with pH adjusted to 2.5 using formic acid.
NOTE: Use 2H2O and deuterated methanol to prepare the modifier solution if the deuterium atoms in the side chains and unprotected backbone amides should be retained for detection by MS.
- Desalting of protein samples
- Prepare an ammonium acetate solution in non-deuterated water at the desired concentration.
NOTE: A concentration less than 100 mM is recommended to avoid a high electric current during electrophoresis and the resulting Joule heating effect. - When necessary, adjust the pH of the ammonium acetate solution to the desired level using formic acid (for pH < 6.8) or ammonium hydroxide (for pH > 6.8).
- Substitute the original buffers of the protein solution with an ammonium acetate solution (prepared in non-deuterated water at the desired concentration; pH adjusted to 7.5 with ammonium hydroxide) through at least five sequential concentrations and dilution steps at 4 °C using a centrifugal filter with a proper MW cutoff.
NOTE: The protein samples to be desalted may be either from prior production procedures (e.g., purification or formulation) or prepared by dissolving the lyophilized powder of protein. The "salts" to be removed from the sample solutions in this step refer in general to all small ions or molecules that are non-volatile. Although these species can be efficiently separated from proteins during the electrophoresis process, this step is recommended to avoid compromising the electrophoretic resolution and thus minimizing the contamination of the mass spectrometer. When protein analytes should be stabilized by specific salts or additives, include them in the BGE. - Determine the protein concentration using a microvolume UV-Vis spectrophotometer.
- Prepare an ammonium acetate solution in non-deuterated water at the desired concentration.
2. Operation of CE-based HDX MS analysis
NOTE: The mass spectrometer used in this approach should be equipped with a mass analyzer with ultra-high resolution, such as a Fourier-transform ion cyclotron resonance (FTICR) or orbitrap, a mass-filter, such as a quadrupole that allows mass-selection of precursor ions for fragmentation, and electron-transfer dissociation (ETD) or electron-capture dissociation (ECD) functions to perform top-down analysis with reliable tandem MS data (ideally isotopically resolved signals of fragment ions).
- Optimization of CE and MS settings
- Perform a pilot MS measurement using a standard electrospray ionization (ESI) source by spraying either the preloaded sample from a metal-coated borosilicate glass capillary (the "static" nanoESI scheme) or the continuously infused sample from a metal emitter to optimize the MS settings for measurement of intact proteins (MS1) and their gas-phase fragments (MS2). Fragment the protein species of interest by mass-selection of the ensemble of its ions in a single charge state, followed by ETD or ECD of the precursor ions.
NOTE: The essential settings include the parameters that affect desolvation, the mass-selection of precursor ions (to avoid interference from other species), and fragmentation efficiency. Both the center and width of the mass-selection window should be increased to match the resulting mass distribution of the analyte ions after HDX. Because the elution window of a protein species in CE typically ranges from 0.5 min to 2 min, assess the fragmentation efficiency based on MS2 scans accumulated over a comparable time window. The optimal values of these parameters are protein-specific; readers are referred to previously published reports for exemplary settings8,14. - Perform a pilot CE measurement using a CE instrument equipped with an optical detector, i.e., a photodiode array (PDA) detector or a UV detector to optimize the CE settings for the separation of the protein species and the migration times, which is equivalent to the HDX reaction times.
NOTE: This step is optional depending on the availability of the optical detector of CE. In the absence of an optical detector, the CE settings can be optimized using CE-MS upon completion of section 2.2, following the instructions described in section 2.3. The essential settings include parameters that affect separation efficiency, peak shapes shown in electropherograms, and elution times.
- Perform a pilot MS measurement using a standard electrospray ionization (ESI) source by spraying either the preloaded sample from a metal-coated borosilicate glass capillary (the "static" nanoESI scheme) or the continuously infused sample from a metal emitter to optimize the MS settings for measurement of intact proteins (MS1) and their gas-phase fragments (MS2). Fragment the protein species of interest by mass-selection of the ensemble of its ions in a single charge state, followed by ETD or ECD of the precursor ions.
- Pre-conditioning of the CE-HDX setup
- Clean the flow-through microvial CE-MS interface with a mixture of 50% methanol, 49% water, and 1% formic acid (v/v) using ultrasonication for at least 30 min at room temperature.
- Upon mounting of the HPC-modified capillary on a CE instrument, rinse the capillary with BGE using the autosampler for 10 min and leave the capillary filled with BGE.
- Obtain a proper length of unmodified fused silica capillary tubing (ID: 50 µm, OD: 360 µm) as the infusion tubing for the modifier solution. Connect the modifier tubing to a gas-tight glass syringe with a blunt tip using a union and proper sleeve, and rinse the tubing with the modifier solution using an infusion pump for at least 10 min.
- Insert the outlets of the HPC-coated CE capillary and modifier tubing, which have been loaded with corresponding solutions, into the cleaned CE-MS interface, as illustrated in Figure 2.
- Advance the syringe for the modifier infusion either manually or with the infusion pump to ensure that the modifier solution reaches the tip of the interface. Mount the assembled CE-MS interface on a nanoESI source housing of a mass spectrometer.
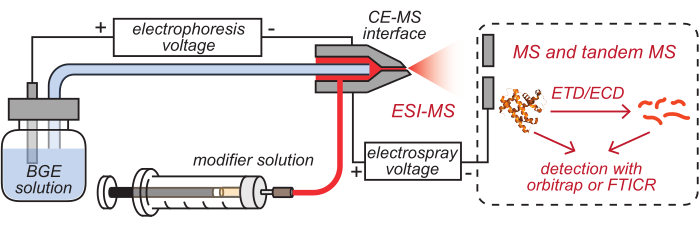
Figure 2: Schematic illustration of the CE-based HDX MS setup. This figure has been modified from8. Abbreviations: BGE = background electrolyte solution; CE = capillary electrophoresis; MS = mass spectrometry; HDX = hydrogen/deuterium exchange; ESI = electrospray ionization; FTICR = Fourier-transform ion cyclotron resonance; ETD = electron-transfer dissociation; ECD = electron-capture dissociation. Please click here to view a larger version of this figure.
- Simultaneous CE separation, HDX reaction, and MS analysis
NOTE: Deuterated BGE is recommended to be used within 1 day after unsealing.- Apply a spray voltage of 3-5 kV to the CE-MS interface.
- Start infusing the modifier solution with the infusion pump at a flow rate ranging from 0.1 to 10 µL/min, and ensure a stable electrospray at the tip of the CE-MS interface.
- Place the sample vial containing the BGE in the autosampler, and use it in step 2.3.4 to acquire blank electropherograms and blank mass spectra.
- Inject the sample solution using the autosampler at 2 psi and for a proper duration to allow the injection of a desired quantity of the sample. Estimate the injection volume using the relationship between injection volume and injection parameters15 defined by equation (1).
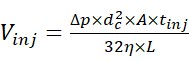 (1)
(1)
Where Vinj is the injection volume, Δp is the pressure of injection, dc is the inner diameter of the capillary, A is the cross-section of the capillary, tinj is the duration of injection, η is the viscosity of the liquid in the capillary, and L is the length of the capillary. - Start the CE separation by applying an electrophoretic voltage of 30 kV and infusion pressure ranging from 0 to 2 psi, and acquire the electropherogram. Meanwhile, start the acquisition of the MS data in the chromatographic mode where the ion current graph is acquired as a function of time, and the corresponding MS scans are not automatically combined into a single spectrum.
NOTE: Proteins undergo spontaneous HDX reaction at the point of contacting 2H2O molecules in BGE during their electrophoretic migration in this step. The optical detection for CE can be used in addition to the MS detection. As on-column detection requires the removal of a certain length of polyimide coating at the outlet end of the fused silica capillary, additional care should be taken to avoid capillary damage during the assembly of the CE-MS interface. - Save the blank electropherogram and mass spectra as references.
NOTE: Blank data are to be used for troubleshooting rather than baseline subtraction. - Place the sample vials containing the desired concentrations of the protein sample solutions in the autosampler. Acquire the electropherograms and mass spectra for the protein samples following steps 2.3.4-2.3.5. Collect an adequate number of MS scans to obtain MS1 spectra of the electrophoretically separated and 2H-labeled protein species.
- Perform tandem MS measurements for the species of interest either after acquiring the MS1 spectra within the same run or in a subsequent, separate run.
- When necessary, adjust the migration times/HDX reaction times by changing the infusion pressure or the length of the CE capillary. If the HDX reaction time must be shorter than the migration time, use the approach described previously8, which employs both deuterated and non-deuterated BGE in the capillary during the CE process.
- Flush the CE capillary with BGE at a pressure of 20 psi for at least 10 min after each measurement.
- Upon completion of the experiments, clean the CE-MS interface and all tubing for storage.
- Acquire a data set of the HDX "endpoint" sample (which can be prepared using approaches described previously6,16) with MS in direct infusion mode.
NOTE: This step is only required when a deuterated modifier solution is used for CE-based HDX.
3. Data analysis
- Analysis of CE data
- Use one of the following plots as the electropherogram to determine the electrophoretic characteristics, including the number of peaks, migration times, and separation efficiency: (a) UV absorbance vs. migration time, acquired by the optical detector of CE instrument (when available); (b) the total ion current (TIC) graph acquired by MS; (c) the extracted ion current (EIC/XIC) graph acquired by MS.
NOTE: EIC/XIC provides the optimal signal/noise ratio (S/N), in general, among the aforementioned formats of electropherograms. It is noteworthy that even in the absence of any instrumental biases, while UV absorbance is proportional to the mass concentration of protein, the MS signal is proportional to the molar concentration. Hence, it is reasonable to observe differences in peak patterns between CE- and MS-derived electropherograms. - Use the area under the curve (AUC) of the peaks shown in the electropherograms for semi-quantitation. For samples involving protein complexes, use the approach described previously17 to deduce the mass concentration data from the TIC/EIC electropherograms.
- Use one of the following plots as the electropherogram to determine the electrophoretic characteristics, including the number of peaks, migration times, and separation efficiency: (a) UV absorbance vs. migration time, acquired by the optical detector of CE instrument (when available); (b) the total ion current (TIC) graph acquired by MS; (c) the extracted ion current (EIC/XIC) graph acquired by MS.
- Analysis of MS data
- Obtain the MS1 and MS2 spectra by combining the MS1 and MS2 scans acquired within the corresponding elution windows, respectively.
- Determine the masses of the intact protein (M (intact protein)) and fragments by either of the following two methods.
- Calculate the average masses of the ions giving rise to the isotopically resolved signal clusters.
- Use the center of the Gaussian-like curves resulting from the fitting of the corresponding isotopic envelopes6.
- Use software such as Biopharma Finder, ProSight18, or MASH Suite19 to generate the mass list of the fragment ions and identify them.
- Analysis of HDX data
- Determine the overall deuteration level of an intact protein species using equation (2).
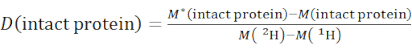 (2)
(2)
where M (2H) or M (1H) are atomic weights of 2H or 1H. The asterisk indicates data of the 2H-labeled sample. - Determine the cumulative protection or cumulative deuteration of backbone-amides of a specific segment.
- For data acquired with a deuterated modifier solution, use equations (3) and (4) to determine the cumulative protection level.
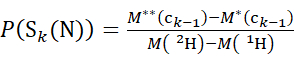 (3)
(3)
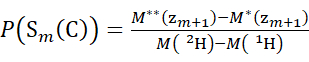 (4)
(4)
Where P(Sk(N)) is the total protection of the N-terminal segment spanning residues 1 through k, P(Sm(C)) is the total protection of the C-terminal segment comprising m residues, M (2H) or M (1H) are atomic weights of 2H or 1H, and M (ci) or M (zi) are the molecular weights of ci or zi ions.
NOTE: The double-asterisk indicates data of the HDX "endpoint" sample. - For data acquired with a non-deuterated modifier solution, use equations (5) and (6) to determine the cumulative deuteration level.
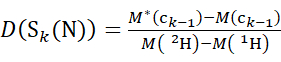 (5)
(5)
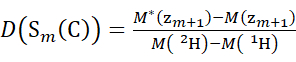 (6)
(6)
Where D(Sk(N)) is the cumulative deuterium uptake of the N-terminal segment spanning residues 1 through k; D(Sm(C)) is the cumulative deuterium uptake of the C-terminal segment comprising m residues.
- For data acquired with a deuterated modifier solution, use equations (3) and (4) to determine the cumulative protection level.
- Determine the deuteration level at a local backbone amide group
- For data acquired with a deuterated modifier solution, use equations (7), (8), (9), (10), and (11) to determine the local protection level.
for data deduced from c-ions
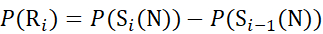 (7)
(7)
for data deduced from z-ions
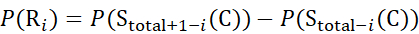 (8)
(8)
Where P(Ri) is the protection of a backbone amide at residue i, and the subscript "total" denotes the total residue number of the protein.
For residue sites where subsequent fragment ions were missing, assign P(Ri) using equations (9) and (10).
for data deduced from c-ions
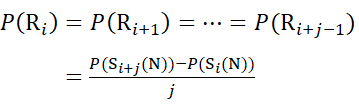 (9)
(9)
for data deduced from z-ions
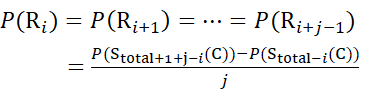 (10)
(10)
Then, determine the deuteration level D(Ri) at a local backbone amide group using equation (11).
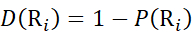 (11)
(11) - For data acquired with a non-deuterated modifier solution, use equations (12), (13), (14), and (15) to determine the local protection level.
for data deduced from c-ions
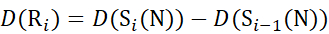 (12)
(12)
for data deduced from z-ions
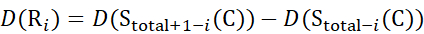 (13)
(13)
Where D(Ri) is the protection of a backbone amide at residue i, and the subscript "total" denotes the total residue number of the protein.
For residue sites where subsequent fragment ions were missing, assign D(Ri) using equations (14) and (15).
for data deduced from c-ions
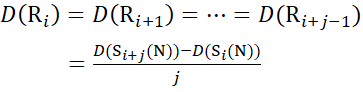 (14)
(14)
for data deduced from z-ions
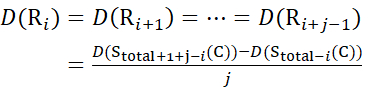 (15)
(15)
- For data acquired with a deuterated modifier solution, use equations (7), (8), (9), (10), and (11) to determine the local protection level.
- Determine the overall deuteration level of an intact protein species using equation (2).
Changing the infusion pressure of BGE allows the adjustment of both separation efficiency and migration time, which is equivalent to the HDX reaction time of the proteins to be separated (Figure 3). A lower infusion pressure results in better separation of CE peaks at the cost of the duration of the experiment (Figure 3A). A longer migration/HDX reaction time results in a higher level of deuteration of the protein analytes (Figure 3B–D). At the HDX timescale of minutes, the deuteration difference should primarily reflect the differing exchange extents at the structurally protected sites instead of the fast-exchangeable sites. According to the trend of the deuteration-time functions shown by either protein species, the migration time difference is unlikely to be the major contributor to the deuteration difference. Indeed, in differential CE-HDX of holo- and apo-myoglobin (Mb)8, the earlier eluted apo-Mb shows a higher deuteration level than holo-Mb, clearly suggesting that the conformational difference is the primary factor determining the measured deuteration difference.
Correction for the deuteration difference introduced by the migration time difference can be made via curve-fitting for the data of the deuteration level vs. HDX time (Figure 3D). The variants A and B of β-lg differ by only two amino acid residues in their sequence (D64G and V118A)20. These variants gave rise to two adequately separated peaks in the EIC-derived electropherogram (Figure 4A). Reproducible separation profiles were obtained from experiments conducted by different operators using different instruments at different facilities (Figure 4A). The resulting distinct mass distributions of ions corresponding to the differentially 2H-labeled variant (Figure 4C) allow mass-selection of each variant using a quadrupole mass filter for the subsequent top-down MS analysis, without interference from the cation-adduct ions of the other variant.
Tandem MS spectra of representative fragment ions are shown in Figure 5. The unique disulfide linkage and conformation of β-lg limit the fragmentation efficiency between Cys82 and Cys176 because additional fragmentation energy is required to cleave the disulfide bonds that enclose this region14, resulting in a lower number and relative abundance of z-ions (C-terminal) than c-ions (N-terminal) (Figure 5A,B). This problem can be solved by combining with disulfide reduction approaches21,22,23,24. While most of the fragment ions produced from β-lg A and β-lg B exhibit a similar extent of deuterium uptake (Figure 5A,B), larger segments that cover the sequence variation sites (represented by ions such as c137) from β-lg A are deuterated to significantly greater extents than β-lg B (132 vs. 119 2H atoms; Figure 5C). These results are in agreement with the CE profile and the crystallography characterization results of these variants. The CE profile indicates higher electrophoretic mobility of β-lg B due to lower structural flexibility. The crystallography characterization results of these variants indicate that small changes in backbone conformation take place in the vicinity of D64G on loop CD (residues 61-67)11.
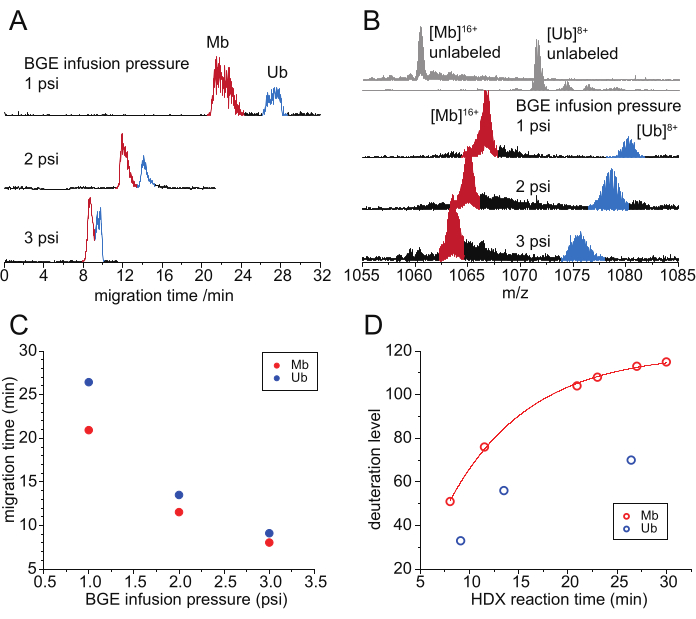
Figure 3: CE-based HDX MS analysis of a mixture of myoglobin and ubiquitin with different HDX reaction times. (A) Electropherograms (EIC-based) of 2H-labeled Mb (red) and Ub (blue) from a mixture, acquired with different BGE infusion pressures. (B) Mass spectra of 2H-labeled [Mb]16+ (red) and [Ub]8+ (blue) acquired with different HDX times. Overlaid on top are reference spectra of unlabeled [Mb]16+ and [Ub]8+ ions (gray). (C) Migration time of Mb (red) and Ub (blue) as a function of BGE infusion pressure. (D) Deuteration level of Mb (red) and Ub (blue) as a function of HDX reaction time (equivalent to the migration time). Data acquired with a CESI 8000 plus capillary electrophoresis system and a Q Exactive UHMR mass Spectrometer. Abbreviations: BGE = background electrolyte solution; CE = capillary electrophoresis; MS = mass spectrometry; HDX = hydrogen/deuterium exchange; Mb = myoglobin; Ub = ubiquitin; EIC = extracted ion current. Please click here to view a larger version of this figure.
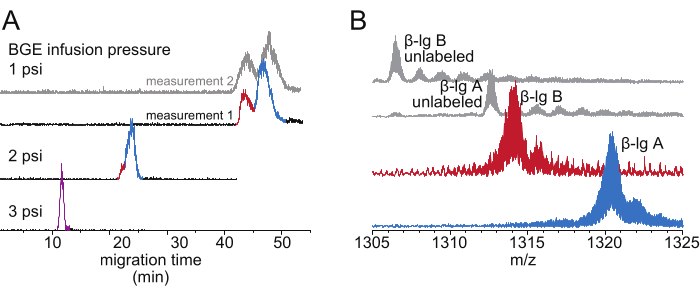
Figure 4: CE-based HDX MS analysis of a natural mixture of β-lg A and β-lg B from bovine milk. (A) Electropherograms (EIC-based) of 2H-labeled β-lg A (blue) and β-lg B (red) from bovine milk, acquired with different BGE infusion pressures. Data acquired with a CESI 8000 plus CE system and a Q Exactive UHMR Mass Spectrometer. Overlaid on top is an electropherogram from a measurement performed at a different facility (gray), with a PA 800 Plus Pharmaceutical Analysis CE System and an Orbitrap Fusion Lumos mass spectrometer. (B) Mass spectra of 2H-labeled [β-lg A]14+ (blue) and [β-lg B]14+ (red) acquired with BGE infusion pressure of 1 psi. Overlaid on top are reference spectra of unlabeled [β-lg A]14+ and [β-lg B]14+ ions (gray). Abbreviations: BGE = background electrolyte solution; CE = capillary electrophoresis; MS = mass spectrometry; HDX = hydrogen/deuterium exchange; Ig = immunoglobulin. Please click here to view a larger version of this figure.
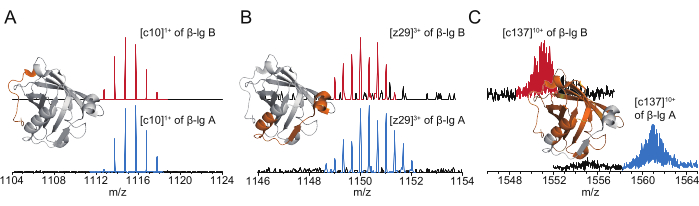
Figure 5: Tandem MS spectra of representative fragment ions produced from β-lg A (blue) and β-lg B (red). (A) c10 ions are abundant and deuterated to similar extents; (B) z29 ions are less abundant and deuterated to similar extents; (C) c137 ions cover the sequence variation sites and are deuterated to significantly different extents in β-lg A and B. The locations of the corresponding segments are illustrated as orange-colored portions of the crystal structure of β-lg B (PDB ID: 5IO5). Abbreviations: MS = mass spectrometry; Ig = immunoglobulin. Please click here to view a larger version of this figure.
