Nodal ligands drive germ layer formation and C&E of zebrafish blastoderm explants
Control explants cut from uninjected wild-type (WT) embryos or those injected with 50 pg of mRNA encoding green fluorescent protein (GFP) remained rounded throughout the culture period (Figure 2A–C) and failed to express markers of mesoderm, endoderm, or neuroectoderm (Figure 3C)20. Together, these indicate an absence of the morphogenesis and germ layer formation that characterize vertebrate gastrulation. However, explants cut from embryos injected with 10 pg of ndr2 mRNA became highly elongated after 8-9 h in culture (Figure 2D). Live time-lapse imaging of these explants by differential interfering contrast (DIC) microscopy revealed that extension onsets at or around 8 h post-fertilization (hpf) (Figure 2F), the same time that C&E morphogenesis begins in intact zebrafish embryos22. Explants cut from MZoep-/- embryos, which lack the essential tdgf1 Nodal co-receptor23, failed to extend in response to ndr2 injection (Figure 2E), demonstrating that Nodal activity is critical for this ex vivo morphogenesis. In addition, whole-mount in situ hybridization further showed that ndr2-expressing explants express markers of neuroectoderm (sox2) and several mesoderm sub-types (tbxta, noto, tbx16) (Figure 2G), as well as endoderm and the embryonic organizer20.
Nodal signaling is not required for neuroectoderm induction by mesoderm
Nodal signaling activity is essential for induction of endoderm and most mesoderm but is dispensable for neuroectoderm specification within zebrafish gastrulae23,24. While uninjected zebrafish blastoderm explants did not differentiate into neuroectoderm (Figure 3C18), explants from embryos injected with 10 pg ndr2 exhibited robust expression of the neuroectoderm marker sox2 in distinct stripes along the long axis of the explant (Figure 2G), indicating that Nodal activity is required for neuroectoderm formation ex vivo. It has long been known that mesodermal tissues can induce neural tissue25,26,27,28,29, including in zebrafish blastoderm explants17. However, it is unclear whether neuroectoderm formation in this explant system requires Nodal signaling directly or whether exogenous Nodal ligands induce mesoderm that then induces neural tissues secondarily.
Chimeric explants containing prospective mesoderm and neuroectoderm portions from two different embryos were generated to test whether Nodal signaling is required tissue-autonomously for neuroectoderm specification ex vivo. The mesoderm portion of each explant was cut from an otherwise WT embryo expressing a mesoderm-specific transgenic GFP reporter, Tg[lhx1a:eGFP]30, injected with a high dose (100 pg) of ndr2 (Figure 3A). The putative neuroectoderm portion of each explant was cut from either a control WT embryo or Nodal signaling deficient MZoep-/- embryo injected only with mRNA encoding the fluorescent nuclear marker H2B-RFP (Figure 3A). Each chimeric explant was generated by combining one blastoderm from each of these two conditions, which were assayed for the expression of tissue-specific markers by whole-mount in situ hybridization at 12 hpf.
The majority of single-embryo explants from embryos injected with 100 pg ndr2 expressed little or no sox2, and expressed markers of mesoderm, including tbxta and the lhx1a:gfp reporter-throughout the explant (Figure 3B,G). Uninjected WT blastoderm (of the type that comprises the prospective neuroectoderm portion of chimeric explants) expressed neither mesoderm markers nor sox2 when cultured as a single explant, indicating a lack of neuroectoderm and mesoderm specification (Figure 3C,H). Single-embryo explants from MZoep-/- embryos similarly lacked expression of both neuroectoderm and mesoderm markers, even when injected with ndr2 (Figure 3D). However, when uninjected WT blastoderms were combined with mesoderm induced by high doses of Nodal ligands, these chimeric explants expressed both mesoderm markers and sox2 robustly (Figure 3E,I). These results demonstrate that, as previously observed17,26,27,29, mesoderm can induce neural fate in cells that would otherwise become non-neural ectoderm. To test whether Nodal signaling is required directly within the prospective neuroectoderm portion of these explants for their neural induction, chimeric explants were created from WT blastoderms injected with 100 pg ndr2 and blastoderms from MZoep-/- embryos (Figure 3J). Despite their inability to receive Nodal signals from the neighboring mesodermal portion, these explants expressed sox2 to a similar degree as WT control chimeras (Figure 3F). This result demonstrates that consistent with intact embryos in which neural tissues are specified in the absence of Nodal activity, Nodal signaling is not required tissue-autonomously for neuroectoderm induction ex vivo.
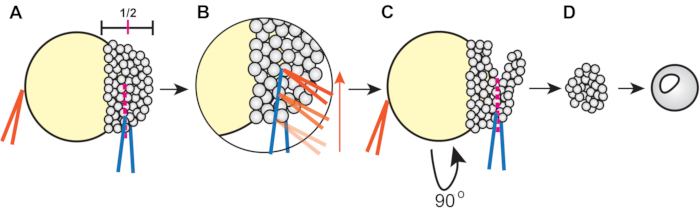
Figure 1: Procedure for zebrafish blastoderm explantation (step 4.3). (A) Hold the forceps in the non-dominant hand (orange) closed against the yolk to stabilize the embryo while pinching the blastoderm at approximately ½ of its height using the forceps in the dominant hand (blue). (B) Run the orange forceps along the edge of the blue forceps gripping the embryo to slice through the blastoderm so that the first cut reaches approximately halfway across the blastoderm. (C) Rotate the embryo 90°, then place the blue forceps inside (but orthogonal to) the original cut and pinch to sever the remaining blastoderm. (D) Allow explanted blastoderm cells to heal in 3x Danieau's solution for approximately 5 min before transferring into explant media. Please click here to view a larger version of this figure.
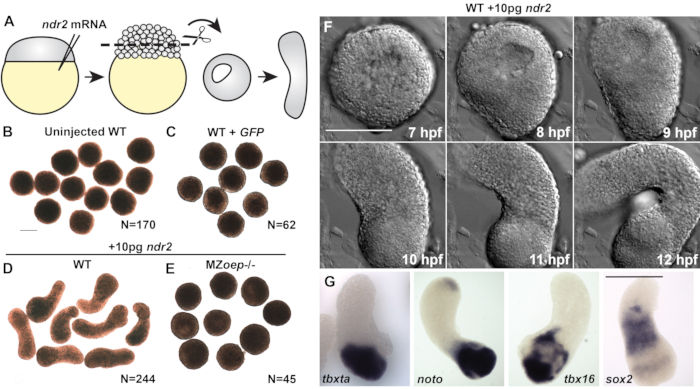
Figure 2 (modified from20): Nodal ligands promote C&E morphogenesis and germ layer formation in zebrafish blastoderm explants. (A) Diagram of injection and explantation of zebrafish embryos. (B–E) Representative bright-field images of live blastoderm explants of the indicated conditions/genotypes at the of equivalent the 2-4 somite stage. N = number of explants from two to four independent trials. (F) Time-lapse DIC series of a representative explant from a WT embryo injected with 10 pg ndr2 RNA. (G)Representative images of the whole-mount in situ hybridization for the transcripts indicated in explants from WT embryos injected with 10 pg ndr2 RNA. Scale bars are 200 µm. Please click here to view a larger version of this figure.
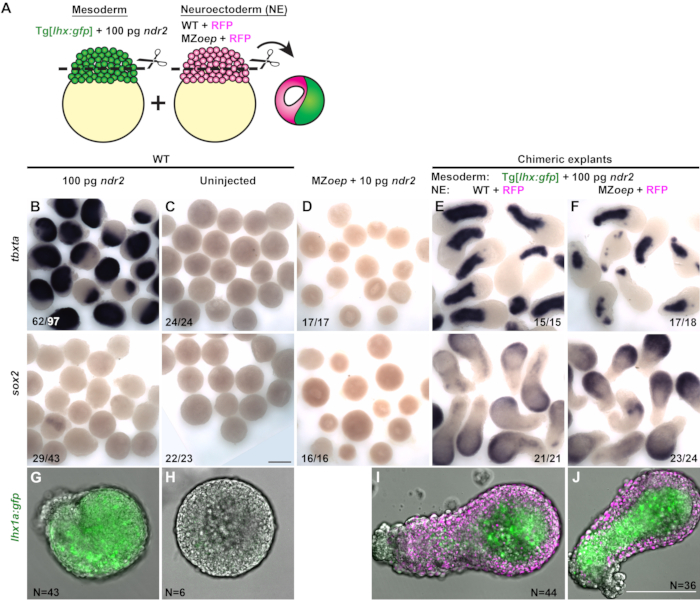
Figure 3 (Modified from31): Chimeric explants reveal that neuroectoderm specification does not require tissue-autonomous Nodal signaling ex vivo. (A) Diagram of the procedure to generate chimeric zebrafish explants. (B–F) Whole-mount in situ hybridization for the mesoderm marker tbxta (top) and neuroectoderm marker sox2 (bottom) in explants from WT embryos injected with 100 pg ndr2 RNA (B), uninjected WT controls (C), MZoep-/- injected with 10 pg ndr2 (D), and chimeric explants containing neuroectoderm portions from WT (E) or MZoep-/- (F) embryos at the equivalent of the 2-4 somite stage. Fractions indicate the number of explants with the phenotype shown over the total number of explants examined. (G–J) Representative images of live Tg[lhx1a:gfp] explants from a single embryo (G–H) or combined with H2B-expressing blastoderms (I–J, magenta) of the conditions indicated at the equivalent of the 2-4 somite stage. N = number of explants from three independent trials. Scale bars are 200 µm. Please click here to view a larger version of this figure.
| Solution 1 | Solution 2 | Solution 3 | Solution 4 | |
| Solution | 3x Danieau's | Egg Water | Explant Media | 0.3x Danieau's |
| Ingredients | 174 mM NaCl | 60 µg/mL sea salts | DMEM/F12 + 2.5 mM L-Glutamine and 15 mM HEPES | 17.4 mM NaCl |
| 2.1 mM KCl | 1 L distilled water | 3% total volume of Newborn Calf Serum or Fetal Bovine Serum | 0.21 mM KCl | |
| 1.2 mM MgSO4.7H2O | 1:200 Penicillin (50 units/mL)-Streptomycin (50 µg/mL) | 0.12 mM MgSO4•7H2O | ||
| 1.8 mM Ca(NO3)2 •4H2O | 0.18 mM Ca(NO3)2 •4H2O | |||
| 15 mM HEPES | Example: | 1.5 mM HEPES | ||
| Distilled water | 4 mL/condition x 9 Conditions = 36 mL | Distilled water | ||
| 1.08 mL Newborn Calf Serum (NCS, aliquoted in -20 °C (3%) | ||||
| 0.18 mL 200x Pen-Strep (PS, aliquoted in -20 °C) (1:200) | ||||
| 35 mL DMEM/F12 |
Table 1.
