There are numerous intracellular events that can be manipulated with actuators responding to light, and that are amenable to bimodal activation with physical and biological light sources. Below are examples employing a photosensing calcium (Ca2+) integrator, light-induced protein translocation, a light-sensing transcription factor, and a photosensitive recombinase. The examples illustrate the feasibility of using bioluminescence to activate various kinds of photoreceptors. The experiments presented were not specifically optimized with respect to light-emitting diode (LED) application, the luciferase chosen, or with respect to concentrations and timing of luciferin application.
Fast light- and activity-regulated expression (FLARE) is an optogenetic system that allows the transcription of a reporter gene with the co-incidence of increased intracellular Ca2+ and light23 (Figure 4A). The presence of Ca2+ is required to bring the protease in proximity to the protease cleavage site accessible only with light stimulation, resulting in the release of the transcription factor. HEK293 cells were co-transfected with the original FLARE components, a dual Firefly (FLuc)-dTomato reporter construct, and a membrane-anchored Gaussia luciferase variant sbGLuc6. In the presence of increased intracellular Ca2+ through the exposure of cells to 2 µM ionomycin and 5 mM calcium chloride (CaCl2), the application of blue LED led to robust expression of the fluorescence reporter compared to cells left in the dark, as well as to the expression of FLuc determined by measuring luminescence upon adding the FLuc substrate, D-luciferin. Similar levels of FLuc expression were achieved with bioluminescence emitted by sbGLuc upon the application of the sbGLuc substrate (CTZ) together with ionomycin and CaCl2. Note that the luciferases used for light activation (sbGLuc) and for reporting the effect of light activation (transcription of FLuc) only produce light with their respective luciferins (CTZ vs. D-luciferin) and do not cross-react.
Different components were combined to generate a light-induced transcription system based on the heterodimerization of cryptochromes23,24 (Figure 4B). CRY2 was fused to a protease while the membrane-bound CIB was fused to the protease cleavage site and transcription factor. Light-induced protein translocation released the transcription factor, leading to the expression of FLuc and dTomato, as shown in Figure 4A. While the presence of the transcription factor component alone resulted in considerable background signal possibly due to spontaneous proteolysis, both physical light (LED) and bioluminescence (CTZ) robustly increased the expression of FLuc as measured in an in vivo imaging system (IVIS).
In another set of experiments, NanoLuc (luciferin: furimazine or hCTZ) was employed for the optogenetic regulation of transcription through the dimerization of CRY/CIB and the photosensitive transcription factor, EL22225,26,27. Figure 5A,B show the schematics of the different components in the dark and light states and the luciferase co-transfected or fused to the light sensor. Various comparisons are shown in Figure 5C. Bioluminescence, induced by adding hCTZ to HEK293 cells expressing the constructs and removing it after 15 min, was more efficient in driving reporter transcription than 20 min of LED light exposure for both CRY/CIB and EL222. For CRY/CIB, an hour of LED exposure was sufficient to reach a level of transcription comparable to 15 min of bioluminescence. In contrast, for EL222, even 60 min of LED were barely half as effective as a brief exposure to bioluminescence. There were no significant differences in the transcription efficacy between the two systems when co-transfected, although the fusion proteins of CRY/CIB were more efficient than those of EL222. For both systems, the fusion proteins led to significantly higher transcription levels than the co-transfected components. CRY/CIB showed consistently higher background levels with vehicle application compared to EL222, which had negligible background transcription. Increasing concentrations of hCTZ alone had no effect on the transcription of the reporter gene.
Photoactivatable recombinases provide a versatile tool for optogenomic manipulations. We tested bioluminescence activation of a photosensitive split Cre recombinase based on the Vivid LOV protein, iCreV28. Figure 6A shows a schematic of the different components, sbGLuc, iCreV, and a lox-stop-lox fluorescence reporter (tdTomato) before and after the application of CTZ. The results from CTZ application relative to controls (no CTZ or LED) are shown in Figure 6B. There is some background expression even in the dark (no CTZ); however, in the presence of CTZ, expression is robustly increased over the background and similar to that induced with LED application.

Figure 1: Light sealed incubator. Cardboard box flap covering the light from the illuminated control panel (top arrow). Light-impermeable cover over the glass door of the incubator (bottom arrow) to protect the cells from light exposure. Please click here to view a larger version of this figure.

Figure 2: Laminar flow hood illuminated by red light. Setup showing a standard laminar flow tissue culture hood being illuminated by red light. Arrow indicates a standard desktop lamp with a red bulb. All manipulations under red light are carried out in an otherwise dark light-tight room. Please click here to view a larger version of this figure.

Figure 3: Light-tight compartments around live-cell imaging microscopes. Two examples of live-cell imaging microscope setups showing the use of either a solid box with plastic drapes only on the front side (left panels: top and bottom) or black drapes all around the imaging setup (right panels: top and bottom). The front sides in both examples remain open and rolled up when not in use (top panels: left and right). The front black drapes are rolled down to prevent any light in the room (e.g., computer screens) from entering the imaging area when performing live-cell bioluminescence stimulation and/or imaging (bottom panels: left and right). Please click here to view a larger version of this figure.
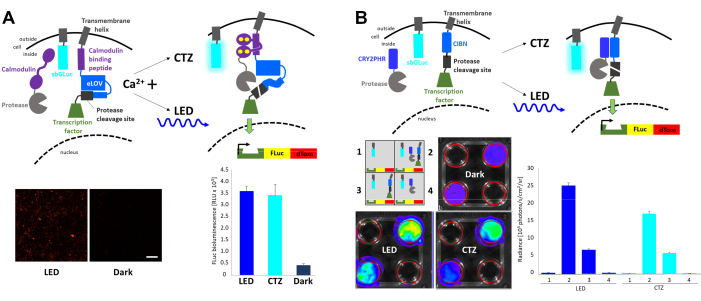
Figure 4: Bioluminescence for integrating intracellular signaling events. (A) Schematics of the FLARE components co-transfected with sbGLuc. In the presence of Ca2+ and the resulting proximity of the protease to the protease cleavage site, either bioluminescence or LED will lead to the unfolding of LOV, exposure of the cleavage site, and release of the transcription factor. Cells were exposed to LED (duty cycle 33%, 2 s on/4 s off for 40 min; 3.5 mW light power, 4.72 mW/cm2 irradiance) or to bioluminescence (100 µM CTZ final concentration for 15 min) or left in the dark. Microscopic images of HEK293 cells expressing the above components after treatment to increase Ca2+ levels and exposure to LED (left). FLuc luminescence measured in a luminometer comparing exposure to LED, bioluminescence (CTZ), or left in the dark (right). (B) Schematics of a non-Ca2+-dependent transcription system co-transfected with sbGLuc. HEK293 cells in 4-well plates were transfected with four different arrangements of components as depicted in the schematic. Plates were exposed to either LED (duty cycle 33%, 2 s on/4 s off for 40 min; 3.5 mW light power, 4.72 mW/cm2 irradiance) or bioluminescence (100 µM CTZ final concentration) by adding CTZ and leaving it on for 15 min; control plates were left in the dark. Transcription of the FLuc reporter was measured in an IVIS. IVIS images of representative dishes are shown on the left; radiance measurements from several replicates baselined to the dark controls are shown on the right. Scale bar = 100 µm. Abbreviations: FLARE = Fast light- and activity-regulated expression; LOV = light-oxygen-voltage-sensing; LED = light-emitting diode; CTZ = coelenterazine; FLuc = firefly luciferase; dTom = dTomato; CRY2 = cryptochrome 2; CRY2PHR = CRY2 photolyase homology region; CIB1 = Ca2+– and integrin-binding protein 1; CIBN = N-terminus of CIB1; IVIS = in vivo imaging system. Please click here to view a larger version of this figure.
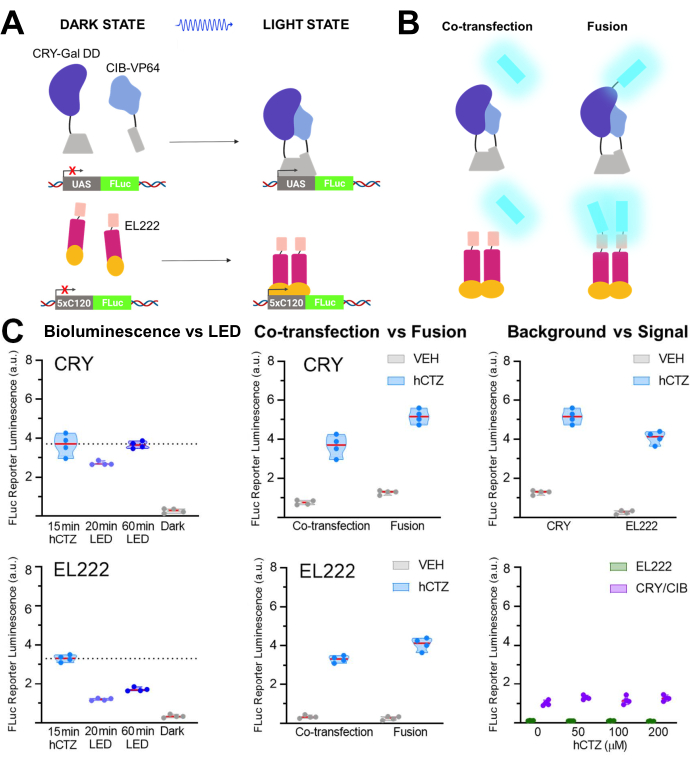
Figure 5: Bioluminescence for driving transcription. (A) Schematics of two photoactivatable transcription systems in their dark and light states. (B) NanoLuc was either co-transfected or fused to the light-sensing moieties as depicted (N-NanoLuc-CRY-GalDD-C; N-NanoLuc-VP16-EL222-C). (C) Comparisons using both systems regarding light sources, construct design, and signal to noise. Cells were exposed to LED (duty cycle 33%, 2 s on/4 s off for 40 min; 3.5 mW light power, 4.72 mW/cm2 irradiance) or to bioluminescence for 15 min (100 µM hCTZ final concentration; except where different concentrations are noted). Dark, plates were left untouched in the incubator between the initial transformation of plasmids and FLuc measurement; VEH, plates were handled the same as those receiving hCTZ, but received vehicle instead. Differences in transcription levels: hCTZ, co-transfected CRY vs. EL222 – not significant; hCTZ, luciferase – photoprotein fusion CRY vs. EL222 – p < 0.005; hCTZ, CRY co-transfection vs. fusion – p < 0.005; hCTZ, EL222 co-transfection vs. fusion – p < 0.01; vehicle, CRY vs. EL222 – p < 0.05. Abbreviations: UAS = upstream activating sequence; LED = light-emitting diode; CTZ = coelenterazine; FLuc = firefly luciferase; CRY = cryptochrome; CIB = Ca2+– and integrin-binding protein; VEH = vehicle. Please click here to view a larger version of this figure.
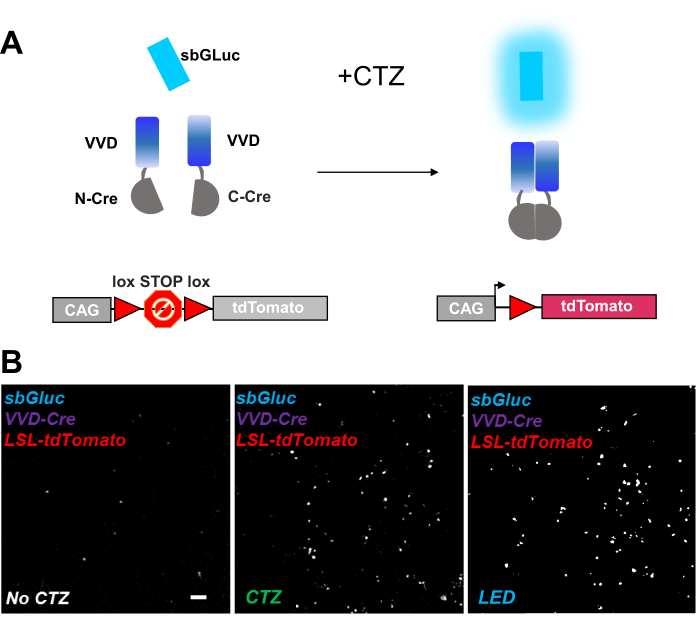
Figure 6: Bioluminescence for optogenomic manipulation. (A) Schematics of bioluminescence-driven optogenomic manipulation using sbGLuc, the split iCreV components, and an LSL reporter cassette, before and after application of light. (B) HEK293 cells were lipofected with plasmids, then kept in the dark. Twenty-four hours later, the cells were treated for 30 min with just medium (no CTZ) or with CTZ (100 µM final concentration) or with LED (duty cycle 25%, 5 s on/15 s off for 5 min; 14.81 mW light power, 20 mW/cm2 irradiance) as a positive control. Microscopic images of tdTomato fluorescence using conditions as indicated. Scale bar = 100 µm. Abbreviations: LSL = lox-stop-lox; CTZ = coelenterazine; LED = light-emitting diode; VVD = Vivid. Please click here to view a larger version of this figure.
Table 1: Bioluminescence activation of photoreceptors. Please click here to download this Table.
Table 2: Guidelines for plating and transfecting cells in different formats. Please click here to download this Table.
Table 3: Ratios of various plasmids for transfection. Please click here to download this Table.
Table 4: Injection routes, volumes, and concentrations of luciferin for in vivo applications (25 g mouse). Please click here to download this Table.
