Three Python scripts are provided here that are compatible with the OT-2 robot, and that perform sample preparation for mass spectrometry proteomics with a single protein standard bovine serum albumin (technical replicates n = 5 digestions) and a detergent-containing human heart lysate sample (n = 5 digestions). Each digest product is partitioned into two peptide clean-up reactions. The number of identified peptide-spectrum matches (PSMs), peptides, and proteins in each run of the BSA and heart samples are shown in Figure 4 and Figure 5. A median of 728 PSMs and 65 peptides were identified with the BSA sample, with 5.2% and 3.2% coefficients of variation (CV), respectively. With the complex heart sample, a median of 9,526 PSMs, 7,558 peptides, and 1,336 proteins was identified in 10 runs with 7.6%, 5.9%, and 3.6% coefficient of variation. A total of 1,935 proteins were identified from 10 runs of the heart sample, and among those, 1,677 proteins were identified in two or more runs. To determine the variability in peptide quantification, the CV of the extracted ion chromatogram (XIC) intensities were calculated for 10 peptides that mapped to a unique protein (Table 2). The variabilities of human (hand-pipetted) vs. robot experimental results on measuring protein concentration were further compared using three protein standard samples with the BCA assay. The average CV (7.57%) of robot BCA assay was found to be lower than the human manual BCA assay (9.22%) (Supplementary Table 1).
The described protocol showed consistent performance over time when the BSA digestion protocol was performed 2 months apart and produced comparable results. The median number of unique PSMs and peptides in Figure 2 are 728 and 65, respectively. The same experiments performed on the OT-2 system 2 months before generated an average of 647 PSMs and 54 peptides (n = 2) (Supplementary Table 2). Longer-term stability may be estimated similarly.
The manual bench processing time (incubation time not included) is calculated between the robot protocol and human processing18 per sample preparation. With the digestion protocol without detergent removal followed by peptide desalting, the manual processing time is 41 min with the robotic system vs. 61 min by hand. With detergent removal, digestion, and peptide desalting protocol, the manual processing time is 54 min with the robotic system vs. 79 min by hand. Therefore, the semi-automated protocol reduces about 20-25 min of hands-on bench processing time per sample. This time reduction becomes considerable when many samples are processed and may be further improved when multiple OT-2 robots are used in parallel.
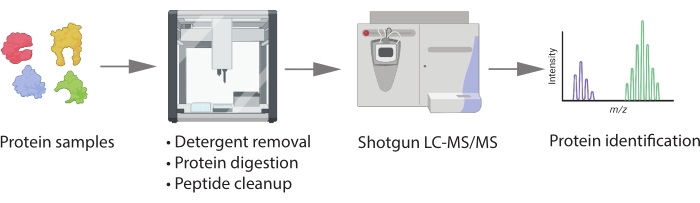
Figure 1: Schematic workflow. Proteins extracted with detergent aid will require processing with an extra step of detergent removal before digestion. Protein samples are digested, and peptides are desalted on the OT-2 robotic system. The peptide digests are injected into a Q-Exactive HF mass spectrometer coupled with a nano-LC. MS spectra are searched against a protein database for protein identification. Please click here to view a larger version of this figure.
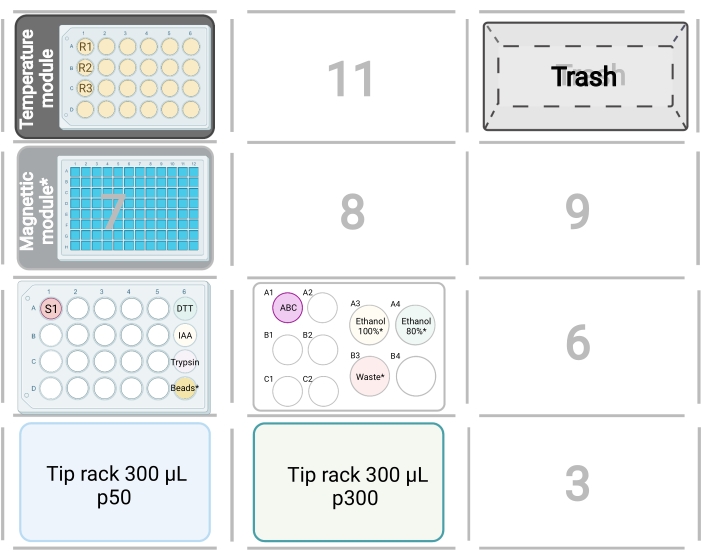
Figure 2: Robot deck set up for protein digestion. The specified positions of tip racks, samples, trash, temperature module, and magnetic module are shown. Asterisks denote labware and reagents that are only required for the digestion protocol with detergent removal steps. Boxes with numbers denote unoccupied deck positions. Please click here to view a larger version of this figure.
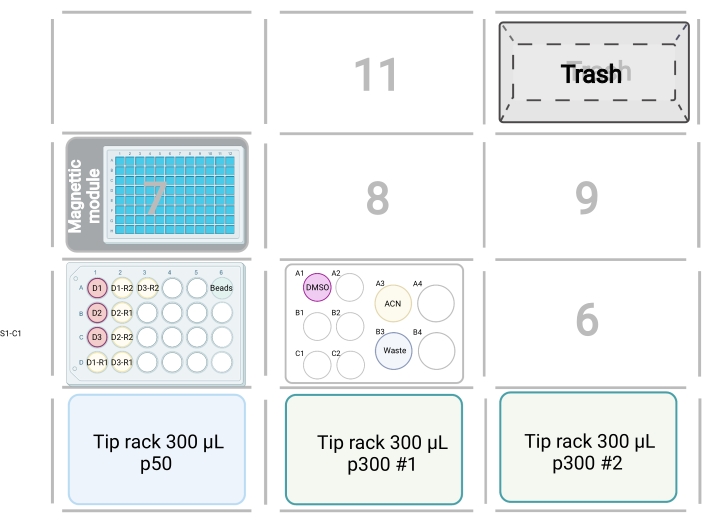
Figure 3: Robot deck set up for the peptide clean-up script. The specified positions of tip racks, samples, trash, and magnetic module are shown. Boxes with numbers denote unoccupied deck positions. Please click here to view a larger version of this figure.
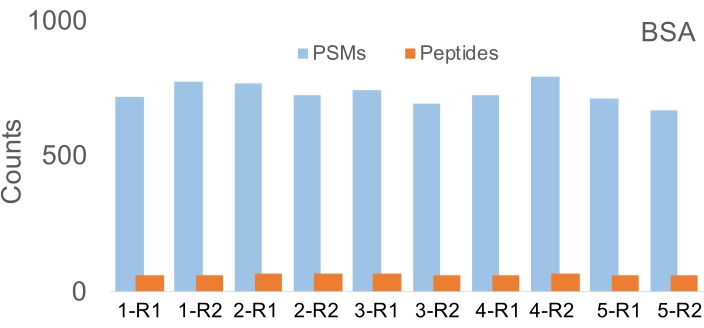
Figure 4: Number of peptide-spectrum matches (PSMs) and peptides detected in the digestions of BSA protein (n = 5). Each digest was split into two for technical replicate peptide clean-ups (R1 and R2). Coefficient of variations: 5.2% for PSMs; 3.2% for peptides. Please click here to view a larger version of this figure.
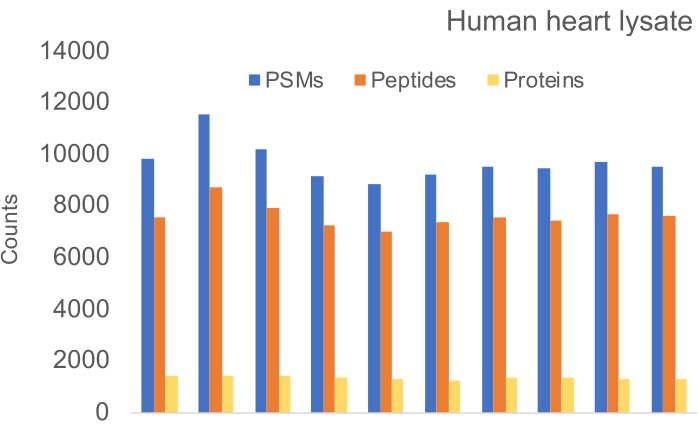
Figure 5: Number of PSMs, peptides, and proteins identified from a human heart lysate. Five digestions were performed with SP3 detergent removal. Each digest was split into two for peptide clean-ups (R1 and R2). Coefficients of variation: 7.6% for PSMs; 5.9% for peptides; 3.6% for proteins. Please click here to view a larger version of this figure.
| Digestion enzyme | Trypsin/P |
| Maximal missed cleavage | 2 |
| Fixed modification | Carbamidomethylation of cysteine |
| Variable modification | N-terminal protein acetylation; oxidation of methionine |
| Peptide length range | 7 – 25 aa |
| Precursor mass tolerance | ± 4.5 ppm |
| MS/MS ions mass tolerance | ± 20 ppm |
| Label-free quantification | LFQ |
| False discovery rate (FDR) for peptide-spectrum match (PSM) | 0.01 |
Table 1: The peptide database (MaxQuant) search parameters.
| Peptide | Protein ID | PEP | Median XIC Intensity | CV | |
| LSTSQIPQSQIR | Q92523 | 7.72E-08 | 1.96E+07 | 6.70% | |
| SEDFSLPAYMDR | P13073 | 9.64E-17 | 8.05E+08 | 7.30% | |
| YLQEIYNSNNQK | P02679 | 2.76E-23 | 9.69E+08 | 7.60% | |
| TDDCHPWVLPVVK | P17174 | 4.51E-14 | 4.60E+08 | 8.60% | |
| VIVVGNPANTNCLTASK | P40925 | 7.90E-29 | 1.17E+09 | 8.70% | |
| DYIWNTLNSGR | O75390 | 1.63E-15 | 1.38E+08 | 8.80% | |
| VSVPTHPEAVGDASLTVVK | P13611 | 1.86E-09 | 6.77E+07 | 9.10% | |
| QVAEQFLNMR | P22695 | 3.25E-08 | 1.09E+08 | 9.30% | |
| NTFWDVDGSMVPPEWHR | Q9UI09 | 2.05E-11 | 4.00E+07 | 9.60% | |
| SASDLTWDNLK | P02787 | 5.29E-11 | 1.92E+09 | 9.80% | |
Table 2: Extracted ion chromatogram (XIC) intensity quantification of 10 peptides.
Supplementary Table 1: Comparison of manual and automated BCA assays. Please click here to download this Table.
Supplementary Table 2: BSA digestion performed 2 months apart from the samples processed in Figure 4. Please click here to download this Table.
Supplementary File 1: A copy of the developed Python scripts. Please click here to download this File.
Supplementary File 2: Method parameters for the liquid chromatography program for LC-MS/MS analysis. Please click here to download this File.
Supplementary File 3: Method parameters for acquiring shotgun proteomics data using a mass spectrometer. Please click here to download this File.
