Commercially available blood samples were used in the present study. At our institution, Institutional Review Board approval is not needed for the use of commercially available blood.
1. Preparation of materials for the comet assay
- Preparation of the microscope slides
- Pour 1% (w/v) normal melting point agarose [dissolved in double-distilled water (ddH2O)] in a 50 mL tube and microwave to dissolve the agarose in the ddH2O. Store at 37 °C to prevent solidification prior to coating slides. Should solidification occur, discard and prepare fresh.
- Pre-coat microscope slides by dipping the slides into the 50 mL tube containing 1% (w/v) normal melting point agarose.
- Wipe the back of the slides quickly after dipping the slides.
NOTE: Failure to wipe the back of the slides properly will increase the background noise of the slides during the analysis step by microscope. - Label the coated slide with a permanent marker on the bottom-right corner of the frosted section (Figure 3A). This shows which side of the slide is pre-coated.
- Allow the agarose to set and dry overnight at room temperature.
- Wrap the dried slides in tissue paper and store them in a box.
2. Preparation of samples
- Cultured cells
NOTE: Firstly, treat cells with the damaging agent(s) prior to starting the comet assay. Then, perform the following.- If the cells are adherent, trypsinize the cells to release them from the cell culture flask or cell culture Petri dishes, at the appropriate confluency of cells. Neutralize trypsin by adding serum-containing media.
- Transfer the cells to a 50 mL tube, centrifuge (e.g., for HaCaTs, centrifuge at 300 x g for 5 min at room temperature), gently remove the supernatant, and add 1 mL of PBS to the cell pellet.
- Perform cell counting.
- Transfer 30,000 cells to a 1.5 mL microcentrifuge tube and centrifuge at 7,607 x g for 5 min at 4 °C.
- Gently remove the supernatant and store the cell pellet on ice in the dark prior to performing the comet assay.
NOTE: Cells should regularly be tested for Mycoplasma contamination prior to performing the comet assay to prevent, among other effects, the formation of artefactual DNA damage and altered DNA damage response, as reported elsewhere23. Centrifugation conditions may be changed, as needed, depending on the cell type used.
- Preparation of cultured cells for repair assay
- Culture cells in the cell culture flask or Petri dishes.
- Wash the cells with 1 mL of PBS twice prior to treating the cells with damaging agents (e.g., for BE-M17 cells, treat with 50 µM of H2O2 for 20 min) on ice to prevent the repair from occurring during the treatment.
- Wash the cells gently with 1 mL of PBS twice to remove any residual damaging agents.
- Re-introduce the cell culture medium and allow the cells to repair for varying durations (e.g., 0 min, 30 min, 2 h, 6 h, 24 h, and 30 h) in a humidified incubator (37 °C, 5% CO2).
- At each time point, collect 30,000 cells in 10% dimethyl sulfoxide (DMSO)-containing cell culture medium and store them at -80 °C.
- Before performing the comet assay, thaw the cells quickly at 37 °C in a water bath and centrifuge them at 7,607 x g for 5 min at 4 °C.
- Remove the supernatant and store the cell pellet on ice prior to performing the assay (i.e., from step 3).
- Preparation of whole blood
NOTE: The following method benefits from (i) being a minimally invasive approach to obtain a blood sample, (ii) not requiring the isolation of PBMC before the comet assay, and (iii) allowing the blood samples (with a volume < 250 µL) to be stored at -80 °C, for up to 1 month (although more recent evidence suggests that longer storage is possible), without the need for cryopreservatives, and with no risk or artefactual damage formation14. Ethical approval, or equivalent, may be needed before obtaining blood samples from patients or animals. Alternatively, commercially available blood samples can be used as in the present study. At our institution, Institutional Review Board approval is not needed for the use of commercially available blood.- Using a pipette, transfer whole blood samples (<250 µL) (Table of Materials) into a collection tube containing a minimal volume containing 0.4 mg of EDTA (per 250 µL of blood).
- Freeze the blood samples at -80 °C before performing the HTP comet assay.
- Thaw stored blood samples (<250 µL) at room temperature, without heating.
- Transfer 5 µL of the whole blood to microcentrifuge tubes prior to performing the comet assay (see step 3).
3. Cell lysis
NOTE: Carry out all the procedures on ice.
- Use 12,000 cells or 2.5 µL of whole blood per gel.
- Prepare 0.6% (w/v) low melting point agarose dissolved in PBS using a microwave, and place in a water bath at 37 °C to prevent it from solidifying.
- Label the frosted end of the pre-coated slides with the investigator's name, date, and treatment information using a permanent marker or pencil.
- Place a chilling plate on a flat bench and insert the two frozen cooling packs into the sliding drawer below the metal surface (as shown in Figure 4)21.
- Place the slides on the chilling plate and allow the slides to pre-chill for 1-2 min before adding the 0.6% (w/v) low melting point agarose-containing cells (step 3.7).
NOTE: Leaving the slides on the chilling plate for more than 1-2 min may cause condensation to form on the slide surface due to ambient humidity. This may make the low melting point agarose gels less stable on the slides. - Disperse the pellet (step 2.2.7) by vortexing. Ensure that all the supernatant has been removed from the pellet. Place the sample tubes (containing the pelleted cells) immediately back on ice.
NOTE: When placing the sample-containing tubes in the centrifuge, put them with the hinge facing outward so that the pellet will be collected on this side of the tube. Sometimes it is difficult to see the pellet, and it is easy to dislodge it while removing the supernatant. Centrifuging with the tube lid in this orientation will enable one to know where the cell pellet will be. - Resuspend the cell pellet with 200 µL of 0.6% low melting point agarose (LMP agarose) and mix by pipetting up and down without creating bubbles. Next, quickly transfer 80 µL of LMP agarose-containing cells onto a chilled slide and quickly place a coverslip onto the gel.
- Allow the gel to set on the chilling plate for 1-2 min.
- Meanwhile, prepare a 500 mL working solution of lysis buffer (Table 1) and pour it into the lysis dish (Figure 2).
- Once the gels have been set, remove the coverslips quickly by gently holding the slide between thumb and forefinger and sliding the coverslip off the gel.
- Place the slides containing samples inside the slide carrier (all the black "dot" marks on slides should be facing in the same direction when they are placed in a carrier) (Figure 3B), and then place the slide carrier inside the lysis dish (Figure 2).
- Close the lid of the lysis dish and keep the lysis dish in the fridge overnight at 4 °C or 30 min at room temperature, whichever best suits the operator's time schedule19.
4. Electrophoresis
- Carefully remove the slide carrier from the lysis dish. Take care not to disturb the gels.
- Gently place the slide carrier in a washing dish pre-loaded with ice-cold ddH2O and leave it for 30 min ensuring that slides are completely covered with ddH2O.
- Insert a frozen cooling pack inside the sliding drawer under the electrophoresis tank to maintain optimal buffer temperature.
- Carefully add ice-cold electrophoresis working solution (Table 1) to the electrophoresis tank and transfer the slide carrier into the electrophoresis tank. Orientate the slides such that their clear parts with the cell-containing gels (i.e., NOT the frosted/labeling ends) point toward the cathode (red electrode).
- Allow the slides to sit in the electrophoresis tank for 20 min so that the DNA relaxes and unwinds. Keep the power supply swtiched off during this step.
- If needed, insert a new frozen cooling pack to maximize chilling.
- Perform electrophoresis for 20 min at 1.19 V/cm, or whatever conditions have been optimized.
NOTE: Optimization of electrophoresis running conditions and volume of buffer is recommended for every laboratory24. Using only a single slide carrier during electrophoresis does not cause any effect of slides on the resistance of the electrophoresis buffer, and the authors did not see a significant effect in the voltage or current when the number of the slides changed. - Switch off the power supply, carefully remove the slide carrier from the electrophoresis tank, and allow it to drain on tissue paper for 30 s.
- Place the slide carrier into the dish containing neutralization buffer (Table 1). Leave it for 20 min.
- Remove the slide carrier from the neutralization dish, place it in the washing dish containing ice-cold ddH2O, and leave it for 20 min.
- Remove the slide carrier from the water and allow the slides to dry in an incubator at 37 °C for 1 h, or at room temperature overnight, or do not dry, depending upon the operator's schedule19.
NOTE: If there is no drying in step 4.11, perform the staining step from 5.2.
5. Propidium iodide (PI) staining
- Transfer the slide carrier to a washing dish containing ice-cold ddH2O to rehydrate the slides and leave for 30 min.
- Place the slide carrier into a staining dish containing 2.5 µg/mL propidium iodide solution.
NOTE: Propidium iodide is light-sensitive, so handle it in a darkened area. It is also toxic. - Close the lid of the staining dish and incubate it for 20 min in the dark at room temperature.
- Transfer the slide carrier to a separate dish, and wash it with ice-cold ddH2O for 20 min.
- Remove the slide carrier from the dish and dry it completely in the dark, either in a 37 °C incubator or at room temperature, depending upon the operator's schedule or preference.
- Once the slides are fully dried, remove them from the slide carrier and store them in a slide box in the dark until ready for image analysis.
NOTE: The slides will remain readable indefinitely and can be re-stained if necessary.
6. Enzyme-modified alkaline comet assay
NOTE: The enzyme-modified alkaline comet assay employs an enzyme treatment step after lysis but before electrophoresis. The activity of the enzyme causes breaks in the DNA at sites that are substrates for the enzyme. Before performing this assay, enzyme concentration and duration of enzyme incubation must be optimized.
- After the cell lysis (step 3), wash slides twice with ice-cold ddH2O for 20 min each.
- Remove the slide carrier from the water and transfer the slides to a tray lined with paper towels.
- Add 80 µL of the enzyme at the optimized concentration (e.g., 3.2 U/mL of hOGG1 for BE-M17 cells, diluted in enzyme reaction buffer) and cover with a coverslip to spread the enzyme over the gel-containing sample.
- Incubate the slides at 37 °C for the optimized duration (e.g., 45 min for hOGG1).
- After incubation, remove the coverslips gently and transfer the slides to the carrier.
NOTE: Do not wash the slides after enzyme treatment; directly perform electrophoresis from step 4.3.
7. DNA inter-strand crosslinks (ICL)-modified alkaline comet assay
NOTE: The concept of this variant of the ICL-ACA is that the presence of ICL in DNA will retard the electrophoretic migration of damaged DNA, induced following exposure to an oxidatively generated insult. In this instance, the shorter the comet tail, the greater the number of ICL25,26,27,28.
- Treat cells with a reagent that induces ICL (e.g., cisplatin; see Supplementary File).
- Expose the treated cells with one of the following agents to induce sufficient strand breaks to create a suitably sized comet tail (~20% tail DNA): hydrogen peroxide (50 µM H2O2 for 30 min), ionizing radiation (2-5 Gy), or Ultraviolet B (UVB) (0.5 J/cm2).
- Additionally, produce a strand break positive control by treating a batch of cells with the same agent and dose, as used in step 7.2 (i.e., no treatment with ICL-inducing agent).
- Centrifuge the cells at 7,607 x g for 5 min, discard the supernatant, wash the cell pellet thrice with 1 mL of PBS, and process as for the alkaline comet assay (steps 3-5).
- Calculate levels of DNA inter-strand cross-linking using the formula below.
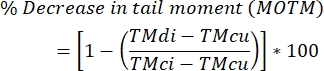
NOTE: MOTM (Mean Olive Tail Moment) is a comet assay endpoint widely used when describing the ICL-modified comet assay, and is defined as the product of the tail length and the fraction of total DNA in the tail (i.e., tail moment = tail length x % of DNA in the tail)29; TMdi: tail moment of samples treated with both crosslinking agent and H2O2 (or other strand breaker inducer); TMcu: tail moment of samples not treated with a crosslinking agent and not treated with H2O2 (no treatment), and TMci: tail moment of samples not treated with a crosslinking agent but treated with H2O2.
8. Comet scoring and data analysis
NOTE: The term "comet" derives from the images of damaged cells when viewed under a microscope after the assay has been performed (Figure 5). Under electrophoresis conditions, DNA in the undamaged cells largely does not migrate, but remains in a spheroid termed as comet "head". However, the presence of strand breaks allows the cell's DNA to migrate out of the head, and form a "tail", thus leading to an appearance like a comet (Figure 5). The more DNA in the tail, the more damage is present.
- Turn on the fluorescence microscope with the PI (red) filter (λ = 536/617 nm) and the comet assay scoring software.
- Add a drop of water using a Pasteur pipette to the gel and cover with a coverslip.
- Place the slides into the fluorescence microscope and "score" the comets.
NOTE: Scoring is a means by which the comets are assessed, to determine the amount of damage present in each comet. Broadly, this can be achieved by using two approaches, according to the user's chosen preference, either by eye (gauging the size of the comets on a scale of zero to four) or by using freely, or commercially available software30. Generally, both approaches assess the size of the comet tail, although a variety of comet-related endpoints can be determined. If using the software, click on the middle of the comet head and wait until the software detects the comet automatically, and then assesses the chosen endpoint (Figure 5). - Score 50 comets per gel and 100 comets per sample (i.e., each sample corresponding to different DNA damaging treatments, or their replicates).
- Replicate the experiments (n = 2) or triplicate the experiments (n = 3).
NOTE: If only n=2 replicate experiments are undertaken, statistical analysis cannot be performed, but if n=3, perform the D'agostino normality test. Most comet assay data does not pass a normality test. In this case, use a nonparametric test (Kruskal-Wallis test with Dunn's multiple comparisons test, and Mann-Whitney tests significance set at p < 0.05).
Optimization of the electrophoresis voltage for the HTP ACA
Human keratinocytes (HaCaTs; Table of Materials) were irradiated with different doses of ultraviolet A radiation (UVA) (5 or 10 J/cm2; Figure 6A), UVB (0.5 or 1 J/cm2; Figure 6B), or treated with 50 µM H2O2 (Figure 6C) to induce damage. Three different voltages of the electrophoresis were tested to determine the optimal voltage for electrophoresis. The results from all three DNA damaging treatments revealed that, while all voltages generated linear dose-responses, the most sensitive response was obtained with 1.19 V/cm. HaCaTs showed the highest baseline DNA damage using 1.19 V/cm during electrophoresis compared to 1 V/cm and 1.09 V/cm (Figure 6A-C). In addition, using 1.19 V/cm, the greatest % tail DNA is seen, following all damaging treatments (Figure 6)31.
Detection of DNA damage in human whole blood using Fpg modified HTP ACA
Human whold blood (Table of Materials) was irradiated with different doses of 10 J/cm2 UVA to induce damage. Four different concentrations of Fpg (1, 2, 4 or 8 U/mL) were used to determine the optimal concentration for enzyme treatment in HTP ACA. The results showed that the optimal levels of DNA damage were revealed with 4 U/mL Fpg (Figure 7A). Representative comet images from UVA irradiated blood samples (Figure 7B).
Detection of DNA ICL in a representative ovarian cancer cell line using the ICL-modified HTP ACA
An ovarian cancer cell line (SKOV-3; Table of Materials) was treated with combinations of 200 µM cisplatin and/or subsequent treatment with 50 µM H2O2 for 30 min on ice. No appreciable damage was noted in the unexposed cells (Figure 8A). Exposure to H2O2 alone generated a significant MOTM (Figure 8B). In contrast, the cells in which ICL were induced showed a decreased MOTM (Figure 8C)28.
Formation and repair of cisplatin-induced DNA ICL in a representative, ovarian cancer cell line
The ICL-modified HTP ACA was used to determine the time course for DNA ICL formation and repair induced by cisplatin in an ovarian cancer cell line (A2780; Table of Materials). The cells were treated with 100 µM cisplatin for 1 h, and then incubated in cisplatin-free media (RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS)) for a subsequent time course. At various time points, the ICL-modified HTP ACA was performed to establish the levels of ICL (Figure 9)28. No ICL were detected prior to cisplatin treatment. However, after a single treatment with 100 µM cisplatin, ICL levels increased significantly, peaking at 12 h, after which levels decreased back to zero after 30 h.
Correlation between DNA ICL and DNA platinum levels
Three ovarian cancer cells were treated with 100 µM cisplatin to induce different levels of DNA-ICL, prior to analysis by the ICL-modified HTP ACA and inductively-coupled mass spectrometry (ICP-MS; see Supplementary File for details). As shown in Figure 10, differing levels of DNA-ICL were induced in the three cell lines, together with differing levels of Pt in DNA. A positive correlation (R2 = 0.9235) was observed between DNA ICL levels and platinum concentrations, indicating the association between DNA platinum levels and the corresponding ICL28.
Base excision repair in Mycoplasma-infected and uninfected BE-M17 cells
Mycoplasma infected and uninfected BE-M17 cells were treated with 50 µM H2O2 for 30 min and incubated with complete medium (Dulbecco's modified Eagle's medium supplemented with 10% (v/v) FBS) for different durations (0 min, 30 min, 1 h, 2 h, 6 h, 24 h, or 30 h) during which cells were allowed to repair. At each time point, cells were collected and frozen at -80 ˚C, in a 10% DMSO-containing medium, before performing the hOGG1-modified HTP ACA (step 6). After 30 min, levels of SB/ALS had decreased to 21% TD (percentage tail DNA) in the uninfected cells, whereas the infected cells showed 49% TD (Figure 11A). After ~15 h, levels of SB/ALS had returned to baseline in both infected and uninfected cells. For the oxidized purines, the uninfected BE-M17 initially showed a small increase in damage, before returning to baseline within 30 h (Figure 11B). In contrast, the infected cells showed a sustained, significant increase in oxidized purines, which remained elevated, and did not return to baseline levels even after 30 h (Figure 11B)23.
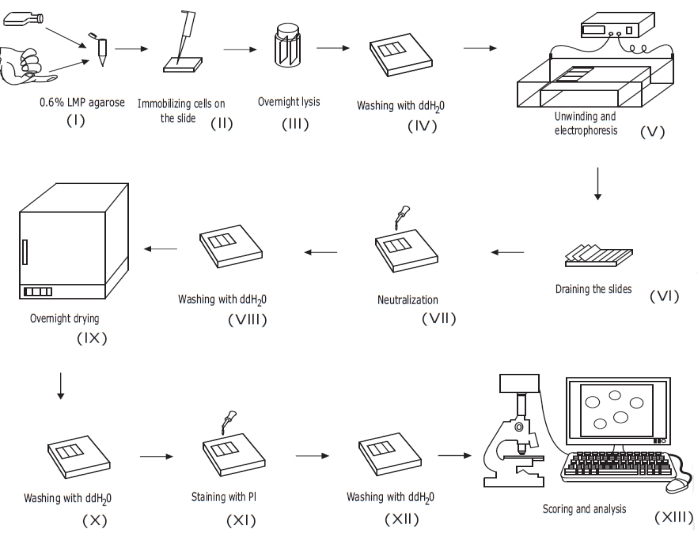
Figure 1: Overview of the conventional alkaline comet assay procedure. (i) A single-cell suspension of cultured cells or a sample of whole blood is mixed with 0.6% (w/v) LMP agarose. (ii) The cell/agarose mixture is applied to pre-coated microscope slides and covered with coverslip until solidified. (iii) The cells are lysed using a high pH lysis buffer overnight, forming nucleoid bodies, before (iv) washing with ddH2O. (v) The cellular DNA unwinds in the high pH electrophoresis buffer. The presence of strand breaks allows the DNA to relax and unwind, and under electrophoresis, the DNA is drawn out of the nucleoid body, forming a tail. The slides are then (vi) drained, dried, (vii) neutralized, and (viii) washed with ddH2O before (ix) being dried overnight. Slides are then (x) rehydrated with ddH2O, (xi) stained, (xii) washed, and finally (xiii) scored and analyzed, typically using fluorescent microscopy and image analysis software. This figure is reproduced from a previous publication20. Please click here to view a larger version of this figure.

Figure 2: The materials comprising the high-throughput comet electrophoresis system. HTP electrophoresis tank, HTP racks, and the dishes for lysis, wash, neutralization, and staining are shown. Please click here to view a larger version of this figure.
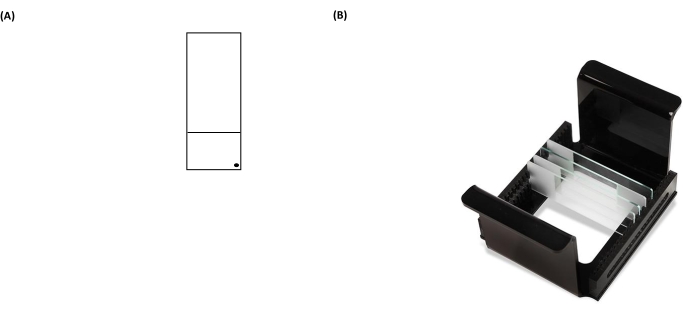
Figure 3: Representative images of a comet assay slide and HTP rack (microscope slide carrier). (A) For correct orientation, the pre-coated face of the microscope slide is recognized by a black dot in the right-hand corner of a microscope slide. (B) The image of the HTP rack illustrates how the slides are kept in a tight vertical orientation, with tabs on the carrier to fix its orientation within the electrophoresis tank. Each carrier can accommodate up to 25 slides. Please click here to view a larger version of this figure.

Figure 4: Representation of the chilling plate with sample slides and freezer packs in place. Please click here to view a larger version of this figure.
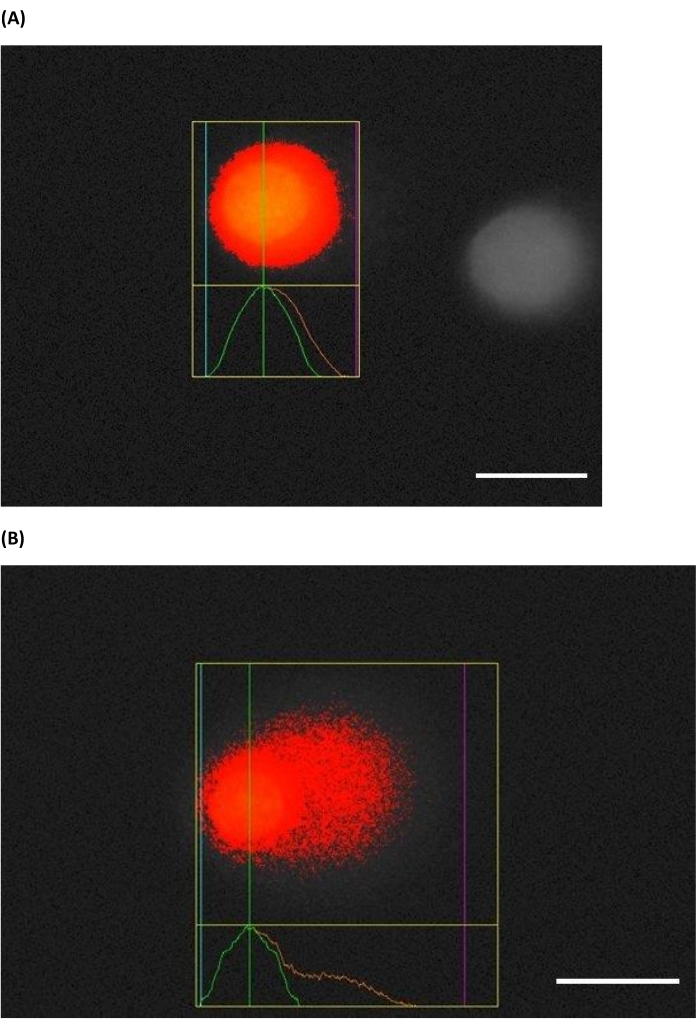
Figure 5: Screenshot of representative comets taken during scoring. HaCaTs (A) without treatment and (B) treated with 1 J/cm2 UVB prior to performing HTP ACA. Most software packages can calculate a variety of comet endpoints, but the most common ones are the % tail DNA (preferred) or tail moment based upon these images (blue: start of the head, green: middle of the head, and purple: end of tail). The scale bar is 10 µm. Please click here to view a larger version of this figure.
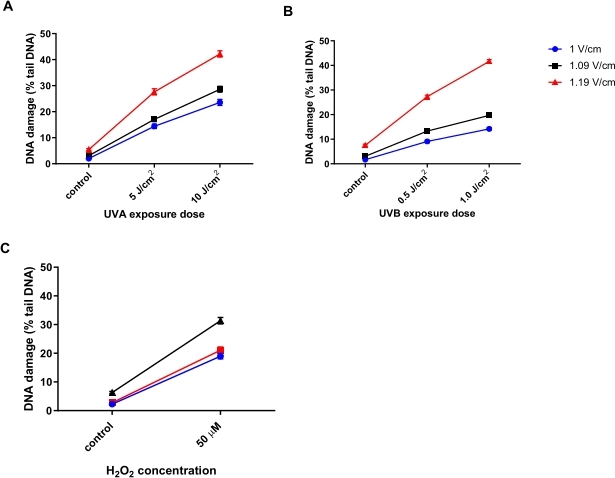
Figure 6: Representative graphs illustrating the effect of electrophoresis voltage on percentage tail DNA, determined using the HTP ACA. Cells were exposed to (A) 5 or 10 J/cm2 UVA, (B) 0.5 or 1.0 J/cm2 UVB, or (C) 50 µM H2O2 prior to the HTP ACA, with the electrophoresis voltage at either 1, 1.09, or 1.19 V/cm. Data represent the mean of 200 determinations from n = 2 duplicate experiments31. Please click here to view a larger version of this figure.
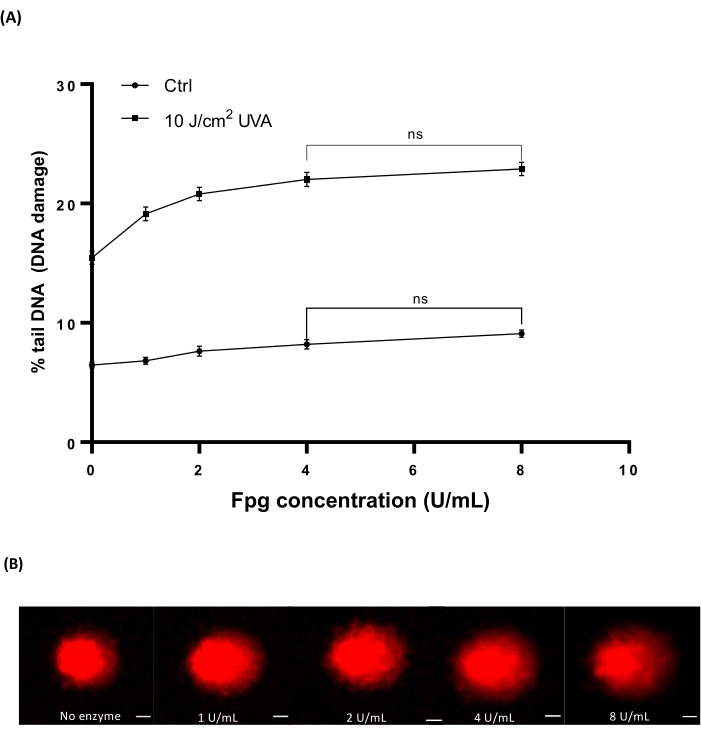
Figure 7: Representative graph and comet images of human blood analysed by the Fpg modified HTP ACA. Human blood samples were irradiated with 10 J/cm2 UVA or sham irradiated ('ctrl') on ice prior to the lysis step. Different concentrations of Fpg (1, 2, 4, or 8 U/mL) were used for the enzyme treatment prior to electrophoresis. (A) Data represent the mean ± SEM of 300 determinations from n=3 experiments. (B) Representative images of comets for each concentration of Fpg in 10 J/cm2 UVA irradiated blood samples. The scale bar is 10 µm. Please click here to view a larger version of this figure.
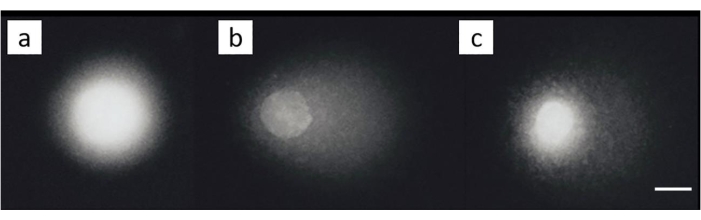
Figure 8: Representative comet images illustrating ICL detection following cisplatin treatment. (A) Control cells without any treatment, (B) cells which were treated with H2O2 (50 µM) only, (C) cells which were treated with H2O2 (50 µM) and cisplatin (200 µM), illustrating the tail to be shorter than in (B), due to the presence of ICL28. The scale bar is 10 µm. Please click here to view a larger version of this figure.
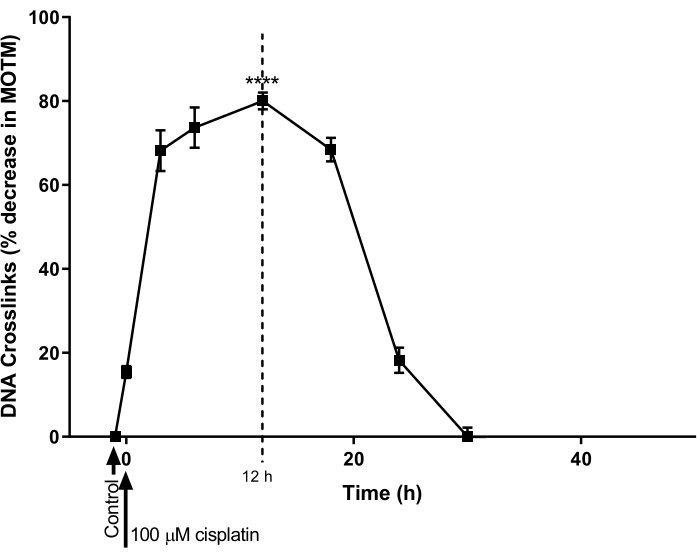
Figure 9: Demonstration of the kinetics of cisplatin-induced ICL formation and repair. A2780 cells were treated with 100 µM of cisplatin in the culture medium for 1 h. The cisplatin-containing medium was then removed, and the cells were cultured for various time points, before analysis by ICL-modified HTP ACA. Data represent mean ± SEM from n = 3 experiments28. **** P < 0.0001. Please click here to view a larger version of this figure.
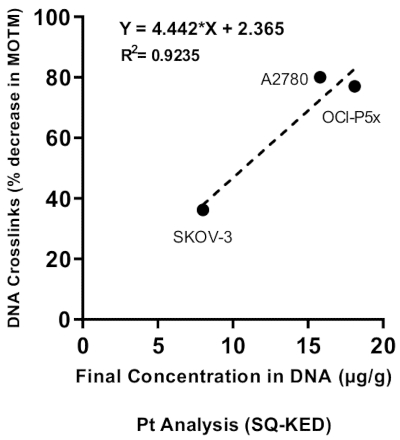
Figure 10: Correlation between DNA ICL and platinum concentration. DNA ICL were determined by the ICL-modified HTP ACA and platinum levels were measured by ICP-MS (with Single Quad-Kinetic Energy Discrimination, SQ-KED), in three ovarian cancer cell lines. R2 = 0.9235. See Supplementary File for ICP-MS methodology to quantify platinum levels in DNA28. Please click here to view a larger version of this figure.
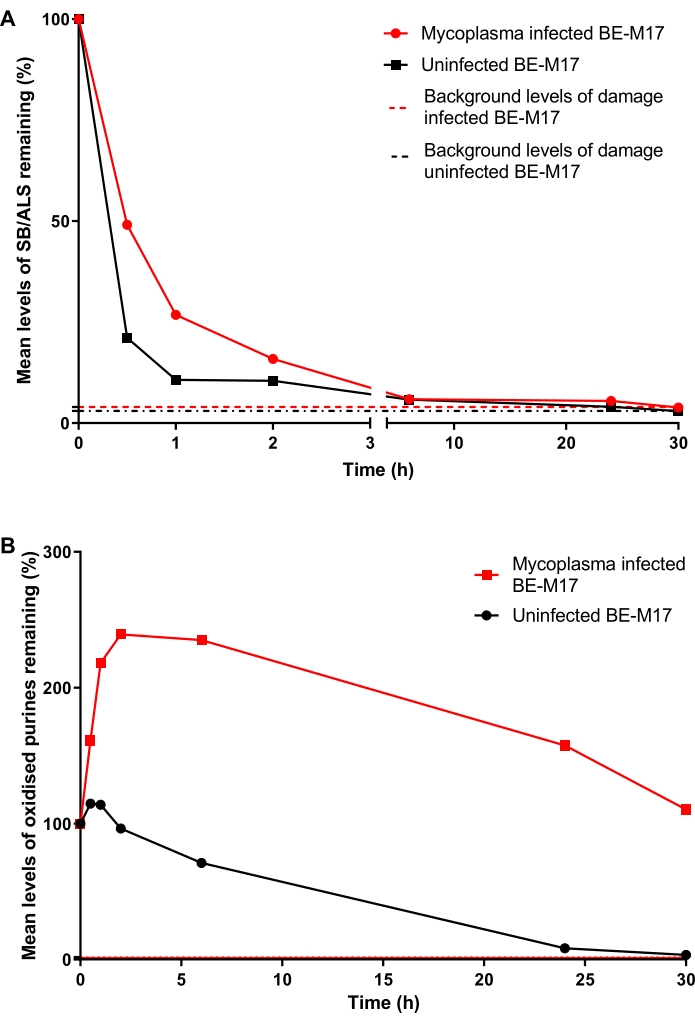
Figure 11: A representative graph illustrating DNA damage and repair, determined by the hOGG1-modified comet assay, in Mycoplasma infected versus uninfected BE-M17 cells. After treatment with 50 µM H2O2 for 30 min, cells were allowed to repair for different durations (0, 30 min, 1 h, 2 h, 6 h, 24 h, or 30 h). The hOGG1-modified HTP ACA was used to measure (A) SB/ALS and (B) oxidized purines in infected (red data points) and uninfected (black data points) BE-M17 cells. Data represent the mean of 200 determinations from n = 2 duplicate experiments. This figure is reproduced with permission from a previous publication23. Please click here to view a larger version of this figure.
| Reagent | Stock Solution | Working solution | |
| Lysis buffer | 100 mM Na2EDTA, 2.5 M NaCl, and 10 mM Tris Base in ddH2O; adjust pH to 10 with 10 M NaOH | 1% Triton X-100 in lysis stock solution | |
| Electrophoresis buffer | 10 M NaOH and 200 mM Na2EDTA in ddH2O | 300 mM NaOH and 1 mM Na2EDTA; pH > 13 | |
| Neutralization buffer | 0.4 M Tris Base in ddH2O; adjust pH to 7.5 with HCl | ||
| Staining buffer | 1 mg/mL propidium iodide | 2.5 µg/mL propidium iodide in ddH2O | |
Table 1: Composition of reagents used in HTP ACA. The stock and working concentrations of lysis, electrophoresis, neutralization, and staining buffers are shown.
Supplementary File. Please click here to download this File.
