Following the experimental procedures shown in Figure 1 and Figure 2, chromaffin cells from bovine adrenal glands were transfected with PH-mNG to label the plasma membrane; A655 was added to the bath solution to detect fusion pore closure; and fluorescent false neurotransmitter FFN511 was loaded in vesicles for detection of release. Next, XY-plane confocal timelapse imaging of FFN511, PH-mNG, and A655 was performed every 20-90 ms at the cell bottom (Z-focal plane ~100-200 nm above the cell membrane). Whole-cell patch-clamp recording and application of a 1 s depolarization from -80 to +10 mV was performed to evoke exo- and endocytosis (Figure 3A–C). This depolarization induced an inward calcium current, a capacitance jump that indicates exocytosis, and a capacitance decay after the jump that indicates endocytosis (Figure 3D, E).
With timelapse XY/Zfixed imaging at the cell bottom, many individual fusion events were observed8,24 after depolarization (Figure 4A), whereas rare fusion events were observed before depolarization. Fusion events induced by the 1 s depolarization protocol were identified as FFN511 spot fluorescence (FFFN) decrease reflecting FFN511 release, accompanied by FPH and A655 spot fluorescence (F655) increase reflecting PH-mNG and A655 diffusion from the plasma membrane and the bath solution into the fusing vesicle (the fusion-generated Ω-profile)15.
After fusion, the Ω-profile generated by vesicle fusion with the plasma membrane may 1) close its pore, termed close-fusion, 2) maintain an open fusion pore, termed stay-fusion, or 3) shrink to merge into the plasma membrane, termed shrink-fusion. Close-fusion was identified as F655 dimming while FPH sustained or decayed with a delay (Figure 4B). Stay-fusion was detected as the sustained presence of both PH-mNG and A655 spots (Figure 4C). Shrink-fusion was detected as parallel FPH and F655 decay accompanied with a parallel size reduction of the PH-mNG spot and the A655 spot (Figure 4D)8,15,16,24.
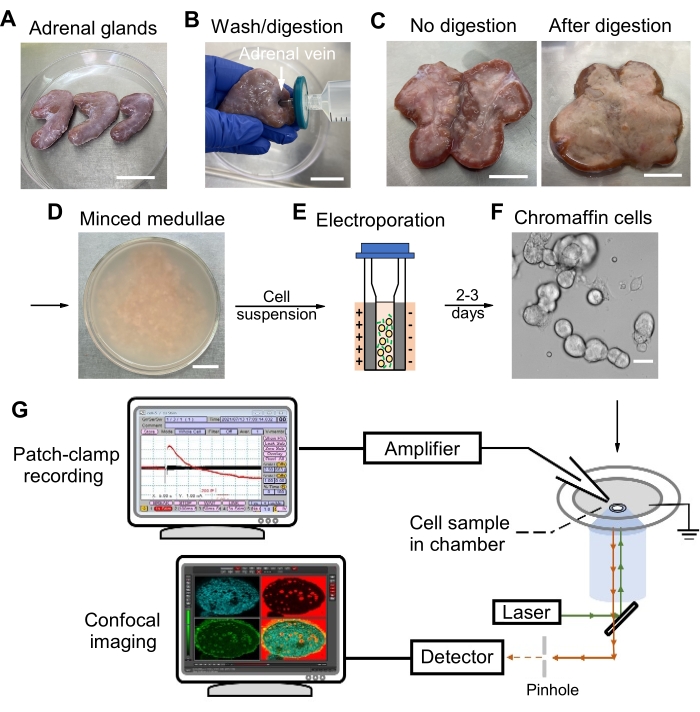
Figure 1: Schematic representation of the experimental protocol. (A, B) Bovine adrenal glands are trimmed with scissors to remove fat tissue (A), flushed with Locke's solution, and digested through adrenal vein (B). (C) The interior of an adrenal gland without digestion (left) or after proper digestion (right). (D) After being washed and digested, the medullae are isolated and minced into small pieces, and chromaffin cells are separated from minced medullae after filtering and centrifuging. (E) Chromaffin cells are electroporated and plated on coverslips for incubation. (F) On days 2-3, check the chromaffin cells under a microscope before experimentation. (G) The cell sample is embedded in the chamber for patch-clamp recording and confocal imaging. Calcium current and capacitance changes are recorded, amplified, and displayed on the monitor. Fluorescence changes upon stimulation are detected and displayed on the monitor. Scale bars = 40 mm (A), 20 mm (B-D), 20 µm (F). Please click here to view a larger version of this figure.

Figure 2: Patch-clamp and confocal setup. (A) On the day of experimentation, the chromaffin cells grown on coverslips are incubated with FFN511 for 20 min. The dye A655 is added to the bath solution. (B) Chromaffin cells on the coverslip are transferred to a recording chamber, and the bath solution with A655 is added to the chamber. (C) After adding a drop of oil on the 100x oil immersion objective, the chamber is mounted onto the stage of an inverted confocal microscope. The tip of the ground wire is immersed in bath solution. The pipette is brought into position after loading with pipette solution and held by a pipette holder, which is attached to a headstage. The headstage is controlled by a motorized micromanipulator. (D) After finding a good cell, move the pipette tip into the bath solution with the micromanipulator to start whole-cell patch-clamp recording and confocal imaging. Scale bars = 10 mm (A), 5 mm (B). Please click here to view a larger version of this figure.
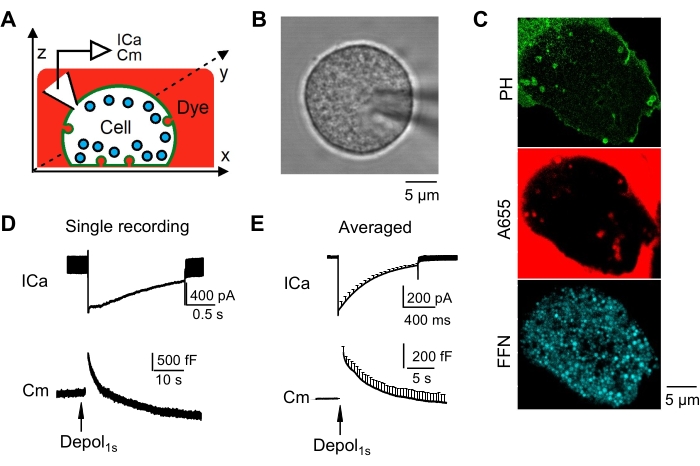
Figure 3: Whole-cell voltage-clamp recordings of calcium currents and capacitance changes induced by depolarization. (A) Setup drawing of chromaffin cells during whole-cell voltage-clamp recording. The chromaffin cell is immersed in A655-containing bath solution (red), and the cell membrane and vesicles are labeled with PH-mNG (green) and FFN511 (cyan), respectively. This image has been modified with permission from 24. (B) A representative image of a patch-clamped chromaffin cell observed by brightfield. (C) Representative images of PH-mNG (green), A655 (red), and FFN511 (cyan) in a cell with good focus at the cell footprint. (D) An example of calcium current and capacitance changes induced by 1 s depolarization from -80 to +10 mV. (E) The averaged traces of calcium currents (ICa) and capacitance (Cm) changes collected from 20 chromaffin cells. This image has been modified with permission from 26. Scale bars = 5 µm (B, C). Abbreviations: ICa = calcium current; Cm = capacitance; Depol = depolarization. Please click here to view a larger version of this figure.
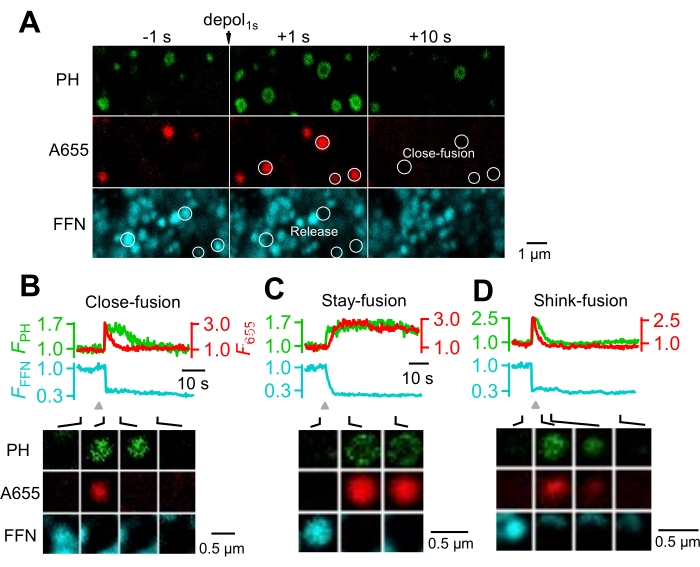
Figure 4: Visualization of fusion events under the confocal microscope. (A) Many fusion spots can be detected with confocal XY-plane imaging of PH-mNG (green), A655 (red), and FFN511 (cyan) at the cell bottom. Fusion was evoked by a 1 s depolarization from -80 to +10 mV (depol1s). FFN spots underwent release and close-fusion (circles). Images before (-1 s) and after (+1 s and +10 s) depolarization are shown. (B) An example of close-fusion. (C) An example of stay-fusion. (D) An example of shrink-fusion. This image has been modified with permission from 24. Scale bars = 1 µm (A), 0.5 µm (B–D). Abbreviations: Depol = depolarization; F = fluorescent intensity. Please click here to view a larger version of this figure.
| Medium/Solution | Description | ||
| Locke's solution | 145 mM NaCl, 5.4 mM KCl, 2.2 mM Na2HPO4, 0.9 mM NaH2PO4, 5.6 mM glucose, and 10 mM HEPES, pH 7.3, adjusted with NaOH | ||
| Enzyme solution | 1.5 mg/mL collagenase P, 0.325 mg/mL trypsin inhibitor, and 5 mg/mL bovine serum albumin in Locke's solution | ||
| Culture medium | DMEM medium supplemented with 10% fetal bovine serum | ||
| Internal solution | 130 mM Cs-glutamate, 0.5 mM Cs-EGTA, 12 mM NaCl, 30 mM HEPES, 1 mM MgCl2, 2 mM ATP, and 0.5 mM GTP, pH 7.2, adjusted with CsOH | ||
| Bath solution | 125 mM NaCl, 10 mM glucose, 10 mM HEPES, 5 mM CaCl2, 1 mM MgCl2, 4.5 mM KCl, and 20 mM TEA, pH 7.3, adjusted with NaOH | ||
Table 1: Details regarding the composition of culture medium and solutions.
