Here, two different sets of results are presented to illustrate classical IVM and ISMic that provide cellular and sub-cellular resolution, respectively. In the first example, neutrophils were purified from a wild-type (WT) mouse, labeled with Cell Tracker Green to stain the cytoplasm, and injected into a transgenic mouse expressing a fluorescent protein targeting the plasma membrane (mTomato mouse, also known as mT/mG33, Figure 1 and Movie 1 and Movie 2). This recipient mouse enabled the visualization of structural features in the ear tissue such as blood vessels, resident cells, and hair follicles (Figure 1C and Movie 1) via IVM-based approach (step 6.8.1). Collagen fibers, revealed via SHG (detected at half the wavelength of the excitation), were arranged in an intricate network in the dermis, where neutrophils were injected. Along the edge of the hair follicles (HF), a layer of epithelial cells (i.e., keratinocytes) was observed. Occasionally, artifacts from residual hairs resulting in a local depression of the skin were visualized (H). The laser-induced injuries were easily visualized due to their strong auto-fluorescence detected in all the channels and by the alterations of the collagen arrangement (Figure 1D). A more complete view of the 3D architecture of the skin and the localization of the injected neutrophils can be appreciated in Movie 1, which portrays a Z-stack of the skin from the outer to inner layers and a 3D volume rendering. Time-lapse imaging showed the neutrophils sampling the ear skin and interacting with the ECM and host tissue (Movie 2). Imaging at this resolution and acquiring a Z stack every 30 s allows performing cell tracking, and measurement of motility parameters (i.e., speed and directionality), but a precise and detailed analysis of membrane remodeling is challenging at this resolution.
In the second example, membrane remodeling was assessed via the ISMic approach (step 6.8.2) using mGFP-expressing neutrophils purified from LyzM-cre mT/mG mice and injected into WT animals (Figure 2A, experimental flowchart). Using the ISMic protocol (Movie 3 and still images in Figure 2B) and upon laser injury, dynamic remodeling of the plasma membrane is observed during migration, and formation of membrane protrusions at the leading edge and the retraction of the rear of the cells are clearly visualized (Figure 2 and Movie 3). The time-lapse sequences highlighted in Movie 3 reveal the complexity of the interactions with the ECM. Indeed, neutrophils migrated through the interstitial space moving either along the fibers or in the spaces between them. Finally, quantitative aspects such as the changes in curvature and the area changes at the front and the back of the cells were quantified for each time point. Using the cell described in Figure 2B as an example, the local dynamics of the plasma membrane were analyzed using an algorithm pipeline (see step 7) based on the identification of 100 boundary points underlying the cell surface (Figure 3A). The changes in the local curvature (Figure 3B) and in the area underlined by plasma membrane protrusions (Figure 3C) were calculated for each boundary point and reported for each time frame as kymographs (Figure 3D,E). Both the front and the back of the cells maintain higher curvature than the side of the cells (Figure 3D); negative area changes (retractions, blue regions) are more apparent at the back of the cells than at the leading edge where the positive area changes are more prominent (protrusions, red regions) (Figure 3E).
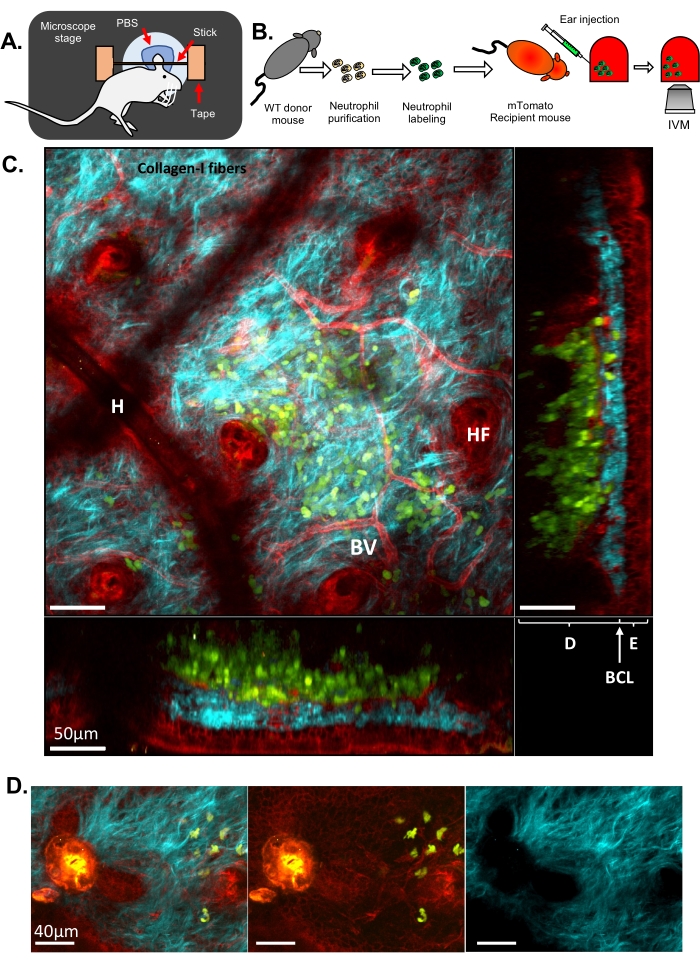
Figure 1: IVM of neutrophils in the mouse ear skin. (A) Schematic drawing of the ear set-up on the microscope stage. (B) Experimental flowchart. (C) Representative image of the ear skin of a mTomato mouse injected with fluorescently labeled neutrophils, with different projections. Hair follicle (HF), Blood Vessel (BV), Epidermis (E), Dermis (D), Basal Cell Layer (BCL), and Hair artifact (due to a residual hair at the skin surface, H). (D) Representative image of mouse skin after a sterile laser injury: blue channel (right), green and red channels (middle), merged image (left). Injury is visible on the left of the image by a strong autofluorescence emission in both green and red channels as well as a disruption of the ECM observed by the formation of a hole in the collagen-I fibers. In C and D, Green: neutrophils; Red: mouse host tissue; Cyan: Collagen-I SHG. Please click here to view a larger version of this figure.
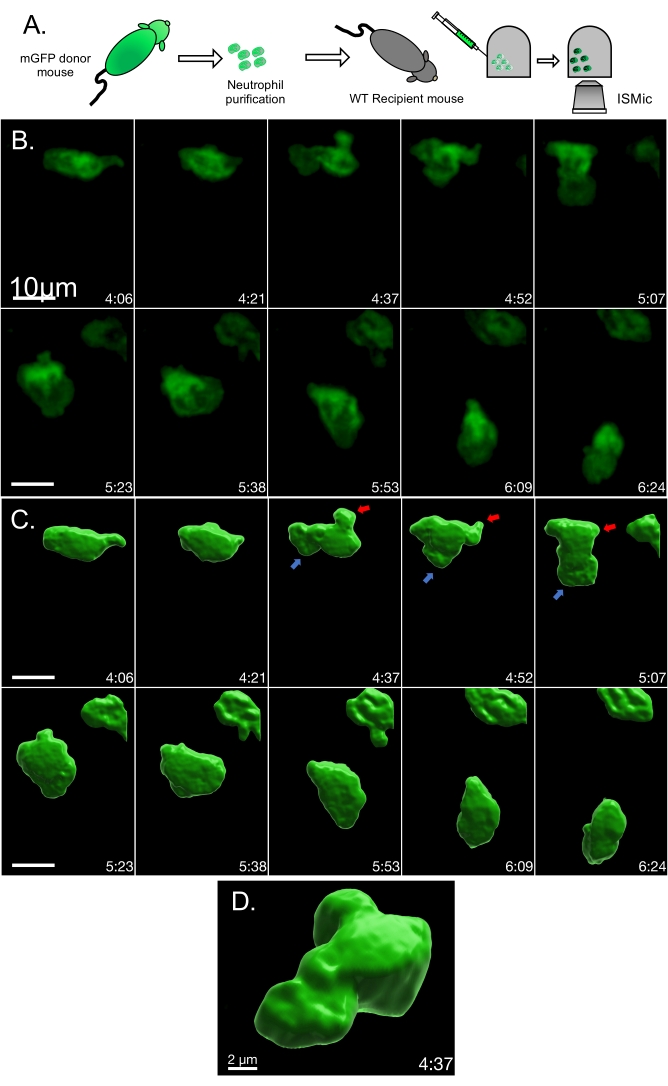
Figure 2: Time-lapse of mGFP neutrophil migrating in a WT mouse ear. (A) Experimental flowchart. (B) Representative still images from Movie 3. (C) Volume rendering of the same cell to visualize membrane 3D organization. The area of high membrane dynamics is illustrated by an arrow (red for retraction and blue for protrusion). (D) Close-up and tilted visualization of the rendered cell volume. Please click here to view a larger version of this figure.
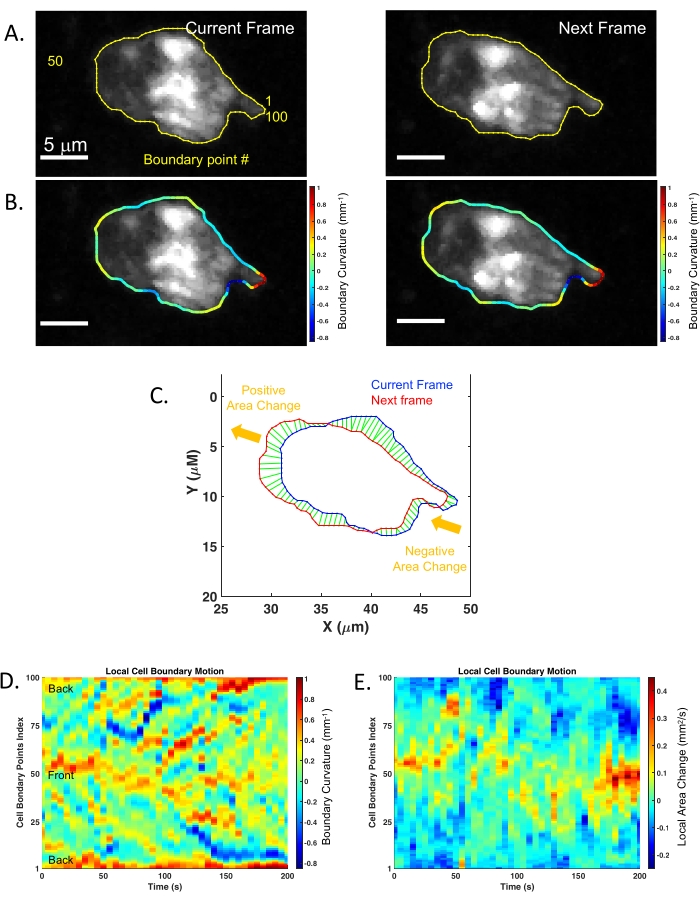
Figure 3: Analysis pipeline for quantification of membrane dynamics collected using ISMic. (A) Cell contour determination and boundary points repartition (100 boundary points) between two consecutive frames for mGFP neutrophil described in Figure 2B. (B) Colored boundaries represent local membrane curvature determined for every boundary point. (C) Local area changes between two consecutive frames (current: blue, and next: red). The green areas represent the tracking results of the local membrane motion. Yellow arrows indicate the general direction of the membrane displacement. (D) Boundary curvature kymograph of the cell over time. The vertical axis represents the boundary points indices where 1 and 100 represent the rear of the cell according to its migration direction. (E) Local area change kymograph reflecting the membrane protrusion (red) and the membrane retraction (blue) of the cell over time. Please click here to view a larger version of this figure.
Movie 1: Green labeled neutrophils visualized in a mTomato mouse ear using IVM. Red: mTomato host mouse tissue. Cyan: Collagen-I SHG; Green: neutrophil. Please click here to download this Movie.
Movie 2: IVM of neutrophils migrating in a mTomato mouse ear skin. Videos are maximum-intensity projections of an image stack acquired for ῀27 min with a frame rate of 5 frames/s. Red: mTomato host mouse tissue. Cyan: Collagen-I SHG; Green: neutrophil. Please click here to download this Movie.
Movie 3: ISMic of membrane remodeling during neutrophil migration in a WT mouse ear. Videos are maximum-intensity projections of an image stack acquired for ῀8 min with a frame rate of 10 frames/s. Red: mTomato host mouse tissue. Cyan: Collagen-I SHG; Green: mGFP neutrophil; Light green: rendered neutrophil. Please click here to download this Movie.
