Western blotting reveals adequate separation of EVs from CCM
To evaluate the effectiveness of SEC for separating EVs from cell culture media, a western blot was run using each individual fraction from the control samples to probe expression of the three canonical EV markers, CD9, CD63 and CD81, as well as GM130 and calnexin18, which were used as negative controls (Figure 3). Albumin expression18 was also probed to ensure that extracellular proteins in the CCM can be adequately separated from EVs. Strong expression of CD9, CD63, and CD81 was observed in fractions 1-4, with little to no expression in fractions 5-8; neither GM130 nor calnexin were observed in any fraction. Albumin was only present in fractions 6-8. Together, these data indicate that vesicles elute predominately into fractions 1-4, leaving fractions 5-8 to contain extracellular proteins. Because of this, fractions 1-4 were combined and concentrated and deemed the EV fraction, while fractions 5-8 were combined and concentrated and referred to as the protein fraction.
Next, additional western blots were run to evaluate the effectiveness of concentrating the EV and protein fractions via ultrafiltration in both control and treated samples. Expression of CD9, CD63, and CD81 was observed in the cell lysates and EV fractions of both control and tunicamycin treated samples, but not in the protein fractions (Figure 4). Tumor susceptibility gene 101 (TSG101) is associated with the endosomal sorting complex required for transport (ESCRT)24 and is commonly used as a marker for exosomes25. This protein was present in cell lysates but not in EV or protein fractions. Additionally, GM130 and calnexin were only observed in the cell lysate samples. Therefore, the data indicates that EVs were effectively separated from cell culture media.
To ensure that the positive signal observed in the EV fractions was coming from EVs released by the cells and not the media itself, the exosome-depleted media underwent the same ultrafiltration and SEC protocol as conditioned CCM and was then analyzed via western blot (Figure 5). Both control and tunicamycin containing medias were processed and cell lysates were used as a positive control on the western blots. No EV markers (CD9, CD63, CD81, or TSG101) were observed in the media samples, but were present in the cell lysates. Albumin expression was observed within the protein fractions and minimal expression in the cell lysates, likely residual from the media. As no signal is observed for any of the EV markers in the EV fractions, these data demonstrate that exosome-depleted media does not contain EVs that mask the signal of cell-derived EVs.
TEM demonstrates expected morphology of EVs
TEM images were taken of both the control and tunicamycin treated EV and protein fractions (Figure 6). Spherical structures can be observed in the EV fractions of both the control and tunicamycin treated samples, indicating the presence of EVs. These structures are not observed in the protein fractions of either sample types, which instead have significant dark staining, indicative of protein in EM imaging.
NTA reveals differences in concentrations in control and treated factions
Particle concentrations of control and tunicamycin treated EV and protein fractions (n = 3) were quantified with nanoparticle tracking analysis (NTA; Figure 7). Figure 7A shows particle concentrations for the control and tunicamycin treated EV fractions, with slightly more particles present in the tunicamycin treatment relative to control. Peak particle concentrations were between 105-165 nm. Figure 7B shows concentrations for particles detected in the protein fraction of both control and tunicamycin treated protein fractions. In the protein fraction, fewer particles overall were detected relative to EVs (109 vs. 1013), and peak concentrations were also smaller, between 75-135 nm. Interestingly, the tunicamycin protein fraction had more particles than the control. The majority of EV-sized particles (approximately 50-200 nm) are observed in the EV fraction of both control and tunicamycin treated cells further supporting the use of SEC as an effective means of EV separation from conditioned cell culture media.
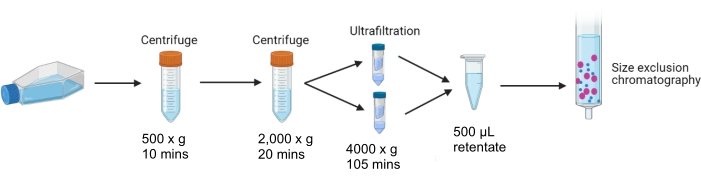
Figure 1: Differential centrifugation and ultrafiltration of conditioned cell culture media. Schematic outlining of the steps of differential centrifugation and ultrafiltration of media collected from cultured cells 24 h after control or 10 µg/mL tunicamycin treatment. Media is centrifuged and concentrated, leaving 500 µL of concentrated retentate suitable for SEC. Please click here to view a larger version of this figure.
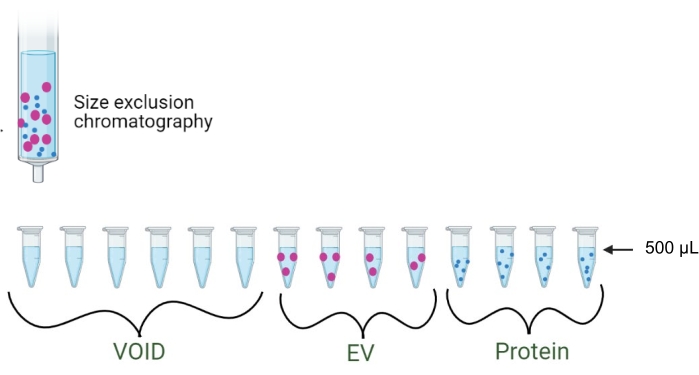
Figure 2: Size exclusion chromatography. Schematic of the separation of EVs and proteins via SEC. The first 3 mL (six fractions of 500 µL each) are discarded as the void volume. Fractions 1-4 elute as EVs, while fractions 5-8 elute as proteins. Please click here to view a larger version of this figure.
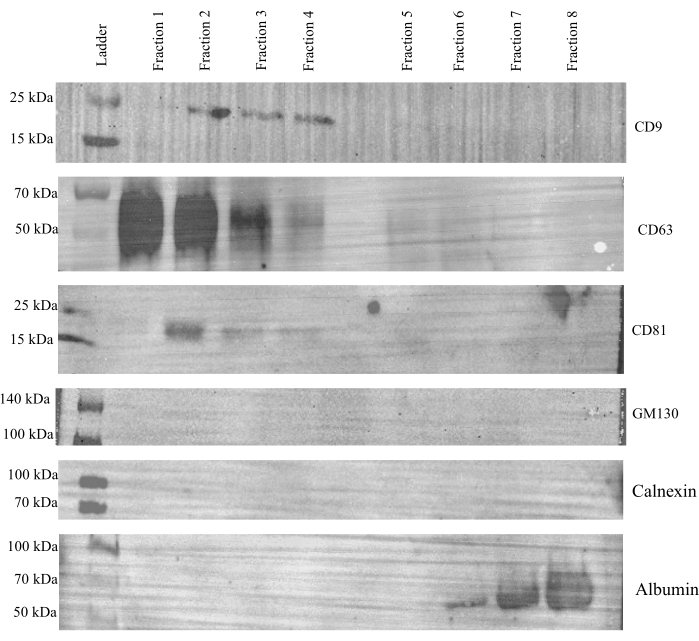
Figure 3: Representative western blot analysis of individual SEC fractions. A volume of 15 µL is used from each individual SEC fraction of the control cells and probed for CD9, CD63, CD81, GM130, calnexin, and albumin expression. CD9, CD63, and CD81 were most strongly observed in fractions 1-4 while GM130 and calnexin were not observed in any fraction. Albumin was only observed in fractions 6-8. Please click here to view a larger version of this figure.
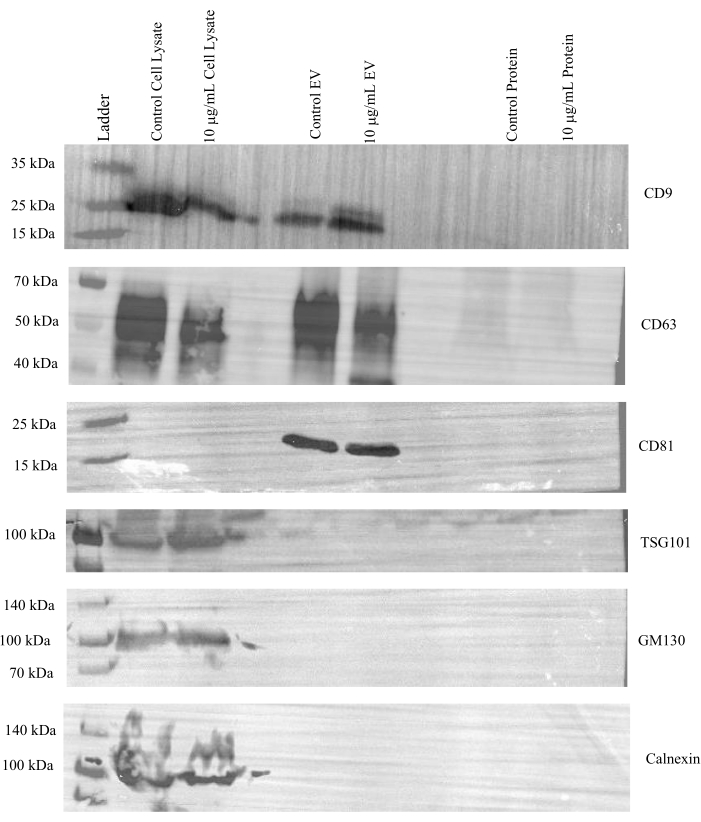
Figure 4: Representative western blot images for canonical EV makers. A final concentration of 20 µg of protein from cell lysates and 15 µg of protein from the pooled and concentrated SEC EV (1-4) and protein (5-8) fractions from control and 10 µg/mL tunicamycin treated cells were probed for CD9, CD63, CD81, TSG101, calnexin, and GM130. All markers were present in the cell lysates. CD9, CD63, and CD81 were observed in all the EV fractions, while TSG101 was not. The negative EV markers calnexin and GM130 were absent in the EV and protein fractions. Control cell lysate refers to lysates of cells that underwent control treatment, while 10 µg/mL cell lysate refers to lysates of cells that underwent tunicamycin treatment. Control EV indicates EVs from control samples. 10 µg/mL EV represents EVs from tunicamycin treated samples. Control protein implies extracellular proteins were from control samples, while 10 µg/mL protein signifies extracellular proteins from tunicamycin treated samples. Please click here to view a larger version of this figure.
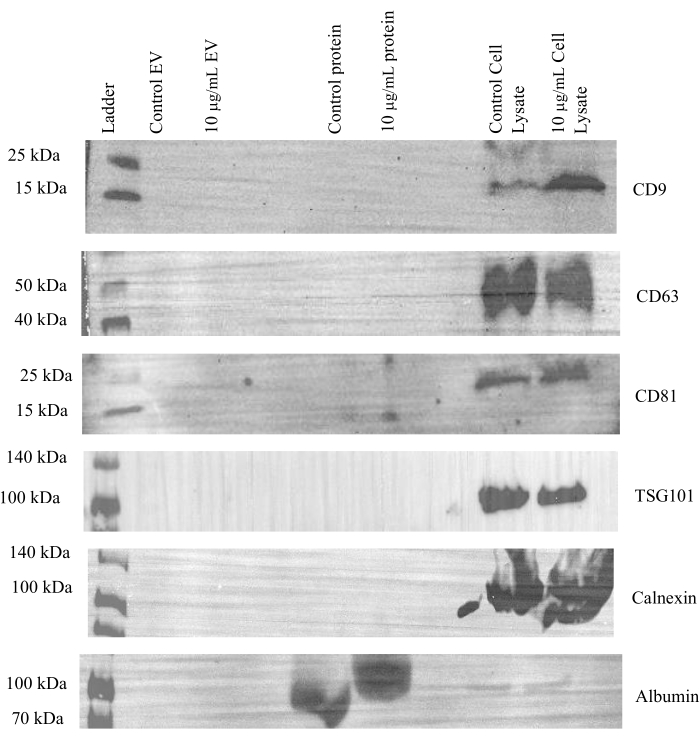
Figure 5: Representative western blot images of exosome-depleted cell culture media. Control and 10 µg/mL tunicamycin exosome-depleted cell culture media underwent SEC and ultrafiltration. A total volume of 15 µL of SEC EV and protein fractions, and 20 µg of protein from cell lysates were probed for CD9, CD63, CD81, TSG101, calnexin, and albumin. No EV markers were observed in the EV or protein fractions but were observed in the cell lysates used as positive controls. Albumin was observed in the protein fractions but not the EV fraction. Control EV indicates EVs from control media samples. 10 µg/mL EV represents EVs from tunicamycin treated samples. Control protein implies extracellular proteins were from control samples. 10 µg/mL protein signifies extracellular proteins from tunicamycin treated samples. Control cell lysate stands for cells in the control treatment that were lysed, while 10 µg/mL cell lysate means lysed cells came from tunicamycin treatment. Please click here to view a larger version of this figure.
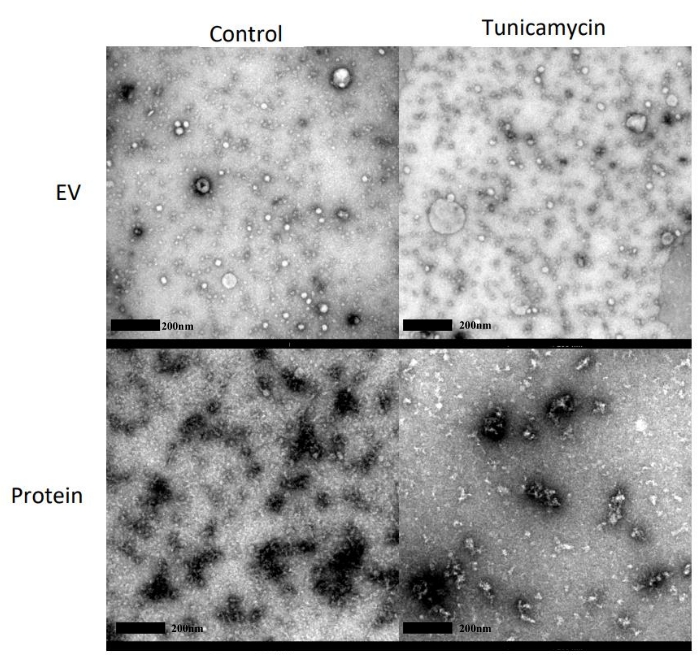
Figure 6: Representative TEM images for morphology of EVs. SEC fractions 1-4 and 5-8 were pooled and concentrated as the EV and protein fractions, respectively, from both control and 10 µg/mL tunicamycin treated cells. EVs are observed in the EV fractions of both the control and tunicamycin samples as small spherical particles less than 200 nm in diameter and are not observed in the protein samples. Scale bars = 200 nm. Please click here to view a larger version of this figure.
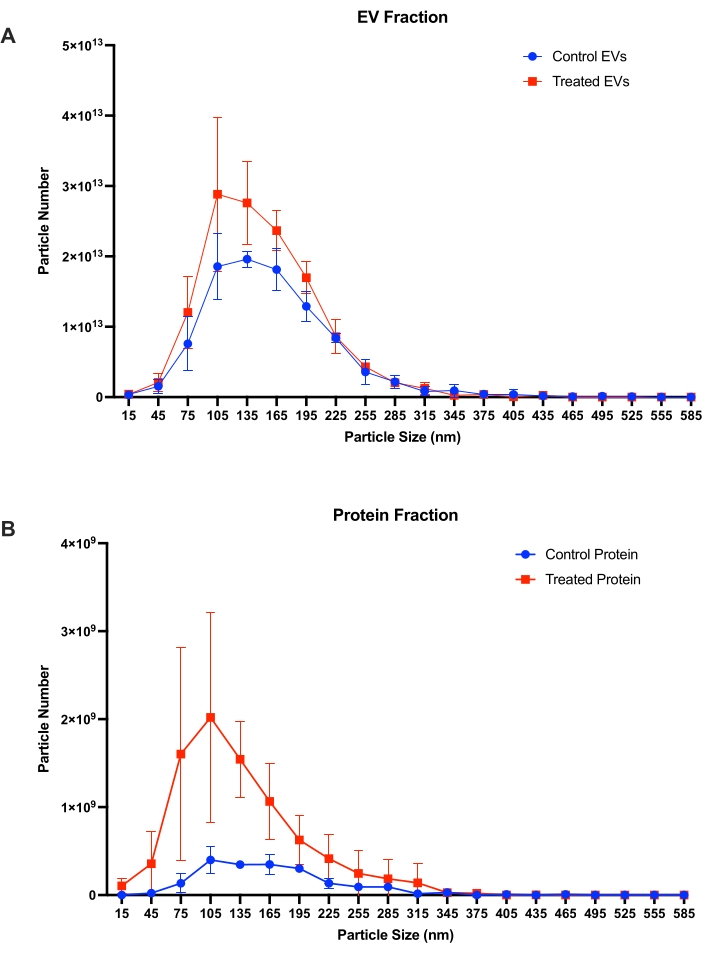
Figure 7: Particle size and concentrations from nanoparticle tracking analysis. Particle size and quantity in (A) EV fractions and (B) protein fractions from control and tunicamycin treated cells (n = 3). Control EVs indicates EV from control samples, while treated EVs represents EVs from tunicamycin treated samples. Control protein implies extracellular proteins from control samples, and treated protein signifies extracellular proteins from tunicamycin treated samples. Error bars are ± one standard deviation. Please click here to view a larger version of this figure.
Supplementary Figure 1: Stain-free image of PVDF membranes from Figure 3. Each PVDF membrane was imaged to visualize successful protein transfer at the conclusion of step 8.3.7. The individual SEC fractions (1-8) were visualized for blots that were later probed for (A) CD9, (B) CD63, (C) CD81, (D) GM130, (E), calnexin, and (F) albumin. Please click here to download this File.
Supplementary Figure 2: Stain-free image of PBDF membranes from Figure 4. Each PVDF membrane was imaged to visualize successful protein transfer at the conclusion of step 8.3.7. Blots were later probed for (A) CD9, (B) CD63, (C) CD81, (D) TSG101, (E) GM130, and (F) calnexin. Control cell lysate refers to cell lysates from control treated cells, while 10 µg/mL cell lysate refers to cell lysates from the tunicamycin treatment. Control EV indicates EVs from control samples and 10 µg/mL EV represents EVs from tunicamycin treated samples. Control protein refers to extracellular proteins from control samples, and 10 µg/mL protein signifies extracellular proteins from tunicamycin treated samples. Please click here to download this File.
Supplementary Figure 3: Stain-free image of PVDF membranes from Figure 5. Each PVDF membrane was imaged to visualize successful protein transfer at the conclusion of step 8.3.7. Blots were later probed for (A) CD9, (B) CD63, (C) CD81, (D) TSG101, (E) calnexin, and (F) albumin. Control EV indicates EVs from control media and 10 µg/mL EV represents EVs from tunicamycin treated media. Control protein refers to extracellular proteins from control media, and 10 µg/mL protein signifies extracellular proteins from tunicamycin treated media. Control cell lysate refers to cell lysates from control treated cells, while 10 µg/mL cell lysate refers to cell lysates from the tunicamycin treatment. Please click here to download this File.
Supplementary Figure 4: Uncropped western blot images used to create Figure 3. Uncropped composite chemiluminescent and colorimetric images of western blots probed for (A) CD9, (B) CD63, (C) CD81, (D) GM130, (E) calnexin, and (F) albumin for each individual SEC fraction (fractions 1-8). Please click here to download this File.
Supplementary Figure 5: Uncropped western blot images from Figure 4. Uncropped composite chemiluminescent and colorimetric images of western blots probed for (A) CD9, (B) CD63, (C) CD81, (D) TSG101, (E) GM130, and (F) calnexin. Control cell lysate refers to cell lysates from control treated cells, while 10 µg/mL cell lysate refers to cell lysates from the tunicamycin treatment. Control EV indicates EVs from control samples and 10 µg/mL EV represents EVs from tunicamycin treated samples. Control protein implies extracellular proteins were from control samples, while 10 µg/mL protein signifies extracellular proteins from tunicamycin treated samples. Please click here to download this File.
Supplementary Figure 6: Uncropped western blot images from Figure 5. Uncropped composite chemiluminescent and colorimetric images of western blots probed for (A) CD9, (B) CD63, (C) CD81, (D) TSG101, (E) calnexin, and (F) albumin. Control EV indicates EVs from control media while 10 µg/mL EV represents EVs from tunicamycin treated media. Control protein implies extracellular proteins were from control media and 10 µg/mL protein signifies extracellular proteins from tunicamycin treated media. Control cell lysate refers to cell lysates from control treated cells, while 10 µg/mL cell lysate refers to cell lysates from the tunicamycin treatment. Please click here to download this File.
| Antibody | Host Species | Dilution |
| CD9 | Mouse | 1 : 500 |
| CD63 | Mouse | 1 : 1000 |
| CD81 | Mouse | 1 : 500 |
| GM130 | Rabbit | 1 : 500 |
| Albumin | Rabbit | 1 : 1000 |
| TSG101* | Rabbit | 1 : 1000 |
| Calnexin* | Rabbit | 1 : 1000 |
| Anti-mouse^ | Horse | 1 : 1000 |
| Anti-rabbit^ | Goat | 1 : 1000 |
Table 1: Antibody dilutions. Dilutions used for antibodies. Stock antibodies were diluted in 5% milk in TBS-Tween. *Represents antibodies that require samples to be run under reducing conditions; ^ represents secondary antibodies.
