The preparation of liposomes based on the inverted emulsion technique is illustrated graphically and schematically in Figure 1.
First, empty (bare) liposomes (~5-50 µm in diameter) that were composed of phospholipid (EPC) and fluorescent lipid (DHPE) were prepared. A bright, far-red fluorescent dye was encapsulated within bare liposomes as a control experiment. Whether a lipid monolayer has successfully formed in the peripheral of the droplet could be determined by observing monolayer lipid droplets incorporating fluorescent dye in the emulsion, as shown in Figure 2A. Successful liposome generation could be verified through the visualization of the thin circular ring, which is the green-fluorescent lipid bilayer (conjugation of lipids with DHPE), under 488 nm laser using confocal microscopy (Figure 2A, right-most panel). To confirm encapsulation of the fluorescent dye, the internal environment of the liposomes should be uniformly fluorescent under a 647 nm laser (Figure 2A; overlay image) due to the far-red fluorescent dye (Table of Materials).
Different forms of encapsulated actin inside the monolayer lipid droplet are shown in Figure 2B. Images from left to right show globular actin (G-actin), filamentous actin (F-actin), actin forming a thin F-actin layer with the addition of VCA-His to the final buffer and Nickel-lipid to the membrane, and actin forming a thin F-actin layer with the addition of VCA-His, cofilin, and gelsolin to the final buffer and Nickel-lipid to the membrane.
Next, purified G-actin and associated F-actin binding proteins were encapsulated within the liposome. Composed of the same membrane components as above (EPC mixed with DHPE), liposomes here were ~5-50 µm in diameter. The formation of liposomes could be visualized by the thin green circular rings shown in Figure 3A,B. Actin polymerization was triggered by allowing the sample to warm to room temperature. As shown in Figure 3A, the reconstituted F-actin networks (with 20% fluorescently labeled actin) inside liposomes are heterogeneous, manifesting as branched network structures of actin filaments. The branched architecture was triggered by the introduction of the Arp2/3 complex, which simultaneously controls the nucleation and branching of actin filaments, along with VCA-His40,41,42,43,44,45.
Finally, an F-actin cortex-biomimicking system was created. A thin, but densely branched F-actin layer was created at the inner leaflet of the bilayer of liposomes22, which could be visualized as a fluorescent shell (Figure 3C). In this case, additionally, VCA-His, cofilin, and gelsolin were encapsulated into liposomes. Nickel-lipids were required in the component of the lipid membrane. VCA-His, a WASP fragment, carries a histidine tag in interaction with the Nickel-lipids of the lipid membrane. In the meantime, it recruits the Arp2/3 complex20,46,47,48. As a result, actin nucleated by the Arp2/3 complex generates an F-actin layer coated along the inner layer of the membrane in the presence of cofilin and gelsolin.
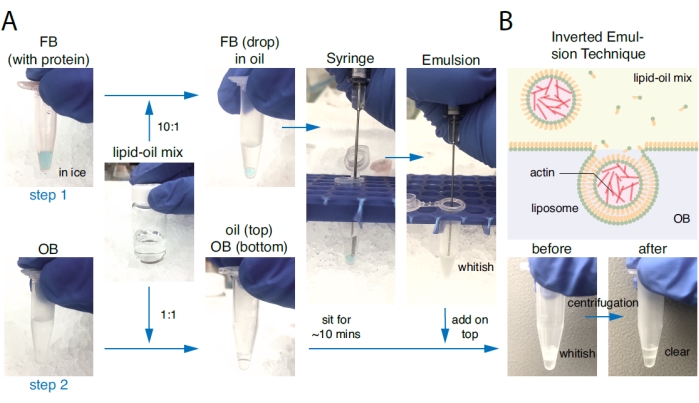
Figure 1: Preparation of liposomes based on the inverted emulsion technique. (A) Liposome preparation consists of two steps. Step one: A droplet of 10 µL of Final Buffer (FB) carrying proteins of interest was added to 100 µL of the phospholipid-oil mixture at a ratio of 1:10 and suspended by gently aspirating up and down with a glass syringe. Solutions containing monolayer lipid droplets became whitish after the back-and-forth aspiration. Step two: In a separate plastic tube, a few microliters of the lipid-oil mixture were placed on top of the same amount of OB and allowed to sit for ~10 min to develop a lipid monolayer at OB/oil interface. The emulsion from step one was poured on top of the oil phase from step two and was centrifuged (100 x g; 15 mins) at 4 °C. After centrifugation, the top oil solution should be clear, and the bottom OB solution (containing liposomes) should be slightly cloudy. (B) Schematics of the liposome preparation from an inverted emulsion. The whitish emulsion on the top contained monolayer lipid droplets that incorporated a branched actin network inside. During centrifugation, monolayer lipid droplets passed through the OB/oil interface to form an outer leaflet such that liposomes were created and accumulated at the bottom of the plastic tube. Please click here to view a larger version of this figure.
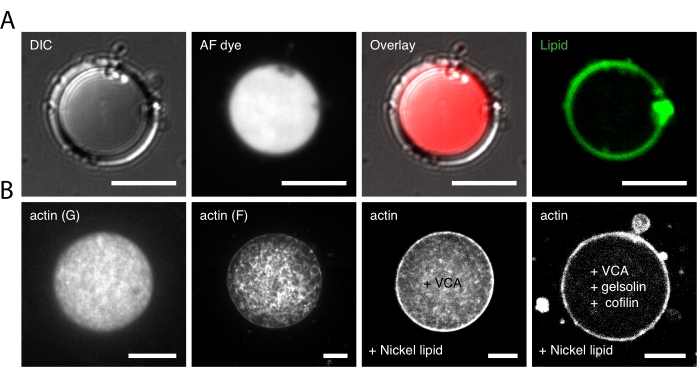
Figure 2: Microscopic images of the monolayer lipid droplet. (A) The monolayer lipid droplet incorporating fluorescent dye in the emulsion. Images from left to right show a monolayer lipid droplet under the DIC channel, fluorescent 640 nm channel, overlay of the DIC and 640 nm channel, and fluorescent 488 nm channel, respectively. (B) Different forms of encapsulated actin inside the monolayer lipid droplet under a fluorescent 561 nm channel. Images from left to right show globular actin (G-actin), filamentous actin (F-actin), actin forming a thin F-actin layer with the addition of VCA-His to the Final Buffer and Nickel-lipid to the membrane, and actin forming a thin F-actin layer with the addition of VCA-His, cofilin, and gelsolin to the Final Buffer and Nickel-lipid to the membrane. Scale bars are 10 µm at 63x magnification. Please click here to view a larger version of this figure.
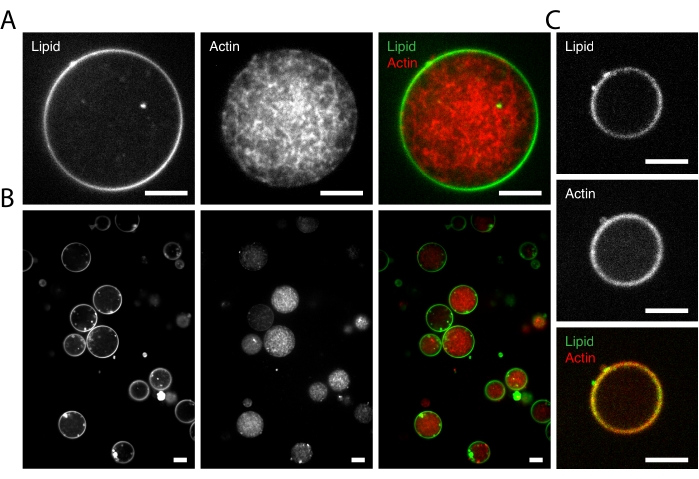
Figure 3: Microscopic images of the encapsulation of actin networks inside liposomes. (A) A liposome encapsulating polymerized branched (Arp 2/3 nucleated) F-actin network. Left to right: fluorescent 488 nm channel (left), fluorescent 561 nm channel (center), overlay (right). (B) A representative result of suspensions of liposomes encapsulating polymerized branched (Arp 2/3 nucleated) F-actin networks. Left to right: fluorescent 488 nm channel (left), fluorescent 561 nm channel (center), and overlay (right). (C) The appearance of the formation of a thin but dense F-actin layer coated along the inner layer of the lipid membrane of a liposome. Top to bottom: fluorescent 488 nm channel (top), fluorescent 561 nm channel (center), overlay (bottom). Scale bars are 10 µm at 63x magnification. Please click here to view a larger version of this figure.
