Due to its intrinsically disordered nature, it is difficult to capture the intricate structural changes in aSyn at physiological pH. HDX-MS monitors isotopic exchange at backbone amide hydrogens, probing the protein conformational dynamics and interactions. It is one of the few techniques to acquire this information at high structural and temporal resolutions. This protocol is broadly applicable to a wide range of proteins and buffer conditions, and this is exemplified by the measurement of the exchange kinetics of aSyn in two different solution conditions: State A and State B8, as defined in steps 2.1.-2.2.
First, a mapping experiment on aSyn was performed, and a peptide coverage map was obtained, as shown in Figure 1. The map covers 100% of the protein sequence and has an average redundancy of 3.79. The 100% coverage value indicates that all the amino acids in the protein were found in the protein digests and will enable a comprehensive analysis of the exchange behavior of aSyn. The redundancy value indicates the number of overlapping peptides. A higher redundancy value increases the structural resolution of the final map, given subtractive flattening of the data for overlapping peptides32.
Using the fast-mixing quench-flow instrument prototype (see Table of Materials), high-quality, millisecond-timescale HDX-MS data on aSyn at pH 7.4 in State A and State B were collected (Figure 2). Following an isotopic assignment in DynamX, "crude" deuterium uptake curves were obtained, as shown in Figure 3A. It shows uptake curves for three peptides selected across each protein domain. The deuterium incorporation over time is displayed. The x-axis is on the millisecond timescale, which aligns with the very fast kinetics of aSyn at physiological conditions. The red shaded region shows the data typically obtained from conventional HDX instruments, with starting measurements from 30 s. Importantly, this cannot be further reduced by pH manipulation for so-called "time-window expansion"; that approach is invalid for the study of intrinsically disordered proteins/regions, as the pH shift will perturb the conformational ensemble of the weakly stable polypeptide. As can be seen here, most of aSyn is fully exchanged by 1 s (Figure 3C). This shows the importance of millisecond HDX measurements for monomeric aSyn as the full kinetic uptake curve for the exchange reaction is captured, which yields the most accurate measurement of the monomer conformations.
HDfleX performed back-exchange correction using the plateau deuterium incorporation. The data points were subsequently fitted according to Equation 1, providing an observed rate constant, kobs, indicative of the solvent accessibility and hydrogen bonding involvement of that particular peptide (Figure 3B).
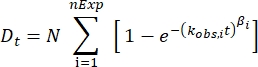 Equation 1
Equation 1
where Dt is the deuterium incorporation at time t, nExp is the number of exponential phases, N is the maximum number of labile hydrogens, kobs is the observed exchange rate constant, and β is a stretching factor30,33.
Following curve fitting, the uptake area under the fitted curve can be calculated by integrating the fitted function describing the uptake curve within the experimental time window. Statistical significance analyses between the uptake area of the two states were performed. First, a global significance threshold for the uptake area was calculated in HDfleX at a confidence level of 95%. Uptake area difference plots were then generated, showing the difference between State A and State B at two levels of structural resolution: peptide resolution (Figure 4A) and amino acid resolution (Figure 4B). The peptide resolution difference plot shows the difference in uptake area between State A and State B for each individual peptide, while the amino acid resolution difference plot shows the difference in uptake area between State A and State B flattened across the entire amino acid sequence of aSyn30,34. Both plots indicate an overall greater deuterium uptake throughout the aSyn monomer in State B compared to State A. This finding can be justified by examining the deuterium uptake plots in Figure 3, where the State B uptake curve is always above the State A uptake curve. Furthermore, it can be seen that the magnitude of the uptake area difference is much higher at the C-terminus. Once again, this can be justified by tracing back to the original uptake curves, where the C-terminal peptides (peptides 124-140 shown in Figure 3) show a much bigger gap between the uptake curves than the rest of the protein. In conclusion, the solution conditions in State B cause an increase in solvent exposure or decrease in hydrogen-bonding network participation throughout the protein but more so at the C-terminus.
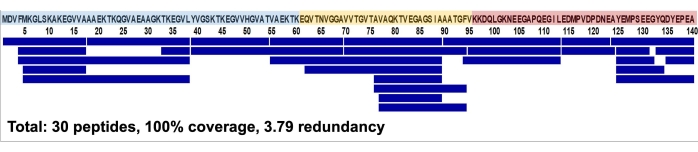
Figure 1: Peptide coverage map of wild-type aSyn with a total of 30 peptides and 100% sequence coverage. The three domains of aSyn are highlighted as follows: N-terminus (blue), non-amyloid beta component region (yellow), and C-terminus (red). Please click here to view a larger version of this figure.
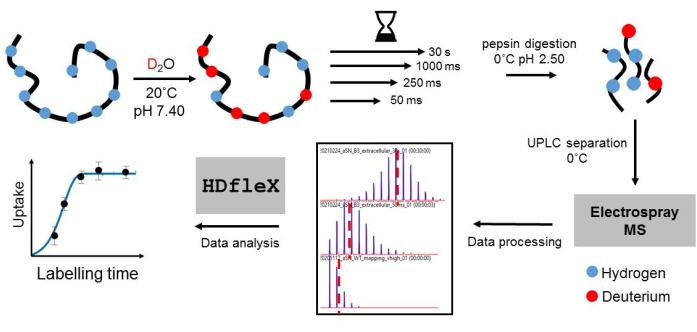
Figure 2: Workflow for a millisecond HDX-MS experiment on aSyn. Please click here to view a larger version of this figure.
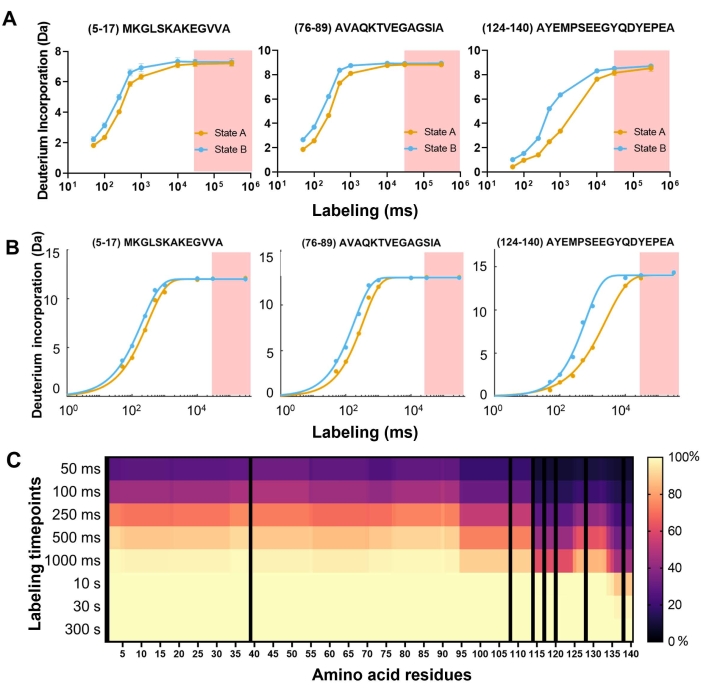
Figure 3: Example uptake plots from three peptides selected across the three domains of aSyn for State A (yellow) and State B (blue). (A) Unfitted and non-back-exchange corrected uptake plot. (B) Fitted and back-exchange corrected uptake plots. The red shaded region represents data obtainable by conventional HDX-MS systems, typically starting from 30 s. Error bars correspond to the standard deviation of the three replicates. (C) Heatmap plot of the percentage deuterium uptake across amino acid sequence per timepoint. The color bar represents the percentage of deuterium uptake. Please click here to view a larger version of this figure.
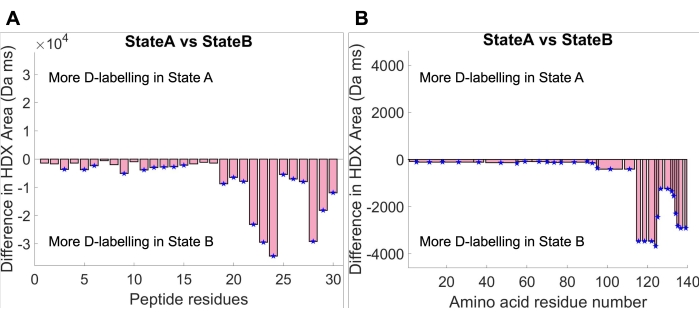
Figure 4: Uptake area difference plots at maximum curve plateau time (14,084 ms). (A) Peptide residue resolution plots show each peptide's uptake area difference between State A and State B. (B) Amino acid resolution plot showing the uptake area difference between State A and State B flattened across the amino acid sequence. Please click here to view a larger version of this figure.
| Mapping Energy Level | Ramp Voltage (V) |
| Low | 20-40 |
| Medium | 25-45 |
| High | 30-50 |
| Very high | 35-55 |
Table 1: Mapping energy levels and corresponding transfer ramp voltages.
