Properly assembled NCPs (Figure 2) were used to make a complex with a recombinant fusion protein of MBP-Polβ-APE1 (Figure 3). To determine the ratio of NCP to MBP-Polβ-APE1 to form a stable complex, we performed electrophoretic mobility shift assays (EMSA) (Figure 4), which showed a singly shifted band of the NCP with 5-fold molar excess of MBP-Polβ-APE1. During the optimization of making this complex, crosslinking with glutaraldehyde was critical to prevent the NCP from falling apart. Initially, assembly of the complex of NCP-Polβ-APE1 was performed in a smaller volume, at approximately 10 µM NCP, using 0.005% final glutaraldehyde concentration. Under these conditions, the sample was overly crosslinked (Figure 5D–E) and resulted in aggregates without discernable individual complexes. Crosslinking at approximately 10-fold diluted NCPs (1.2 µM NCP) resulted in significantly reduced aggregation and improved particle stability (Figure 5A–C). Figure 6 illustrates that both methods shown in Figure 1 can be combined with the preparative gel improving the quality of the NCPs when the NCP contains a significant amount of high molecular weight (HMW) aggregates (Figure 6A), followed by the purification of the complex via size exclusion. Although this showed great success in the preparation of the sample (Figure 5B–C) and grids, yielding stable particles (Figure 6D), the suboptimal formation of the complex (Figure 6A) yielded a 3D map of the NCP alone (data not shown). These results show that both methods (size exclusion and preparative gel) can be used independently to generate stable complexes (Figure 7A–C). Indeed, data have been collected from both grids and show almost identical 2D classes (Figure 7D) and 3D maps (Figure 7E) at approximately 3.2 Å resolution.
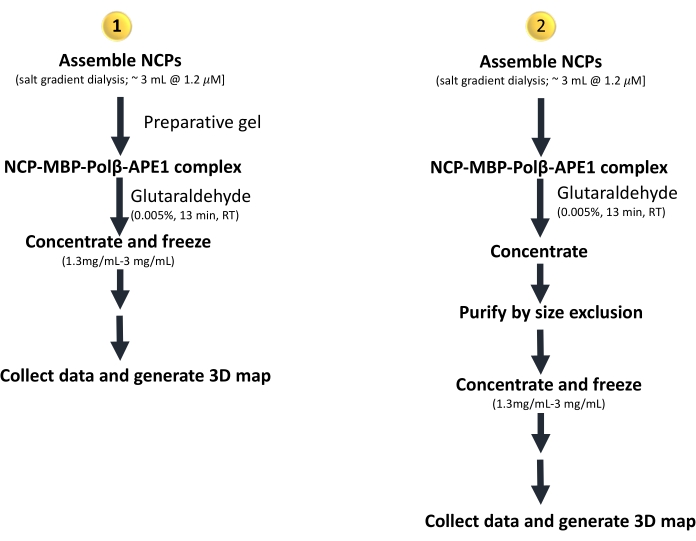
Figure 1: Workflow of the two methods presented in this protocol to prepare and freeze an NCP-Polβ-APE1 complex via 1) preparative gel and 2) size exclusion. Although not shown here, these methods can be combined (Figure 6), starting with (1) and purifying the concentrated complex from (1) using a sizing column. This will require doubling the starting material of NCPs. Please click here to view a larger version of this figure.
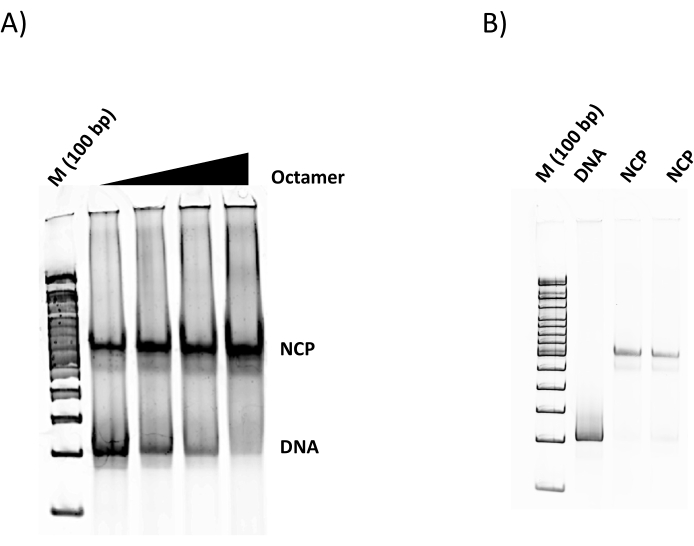
Figure 2: Representative NCP reconstitutions. (A) DNA was titrated with increasing amounts of histone octamer at ratios of 1:0.9, 1:1.1, 1:1.2, 1:2, DNA:octamer. (B) Duplicate large-scale assembled NCPs (1:2, DNA:octamer). Reconstitutions were electrophoresed in a 6% nondenaturing polyacrylamide gel, followed with sybr green I staining. Please click here to view a larger version of this figure.
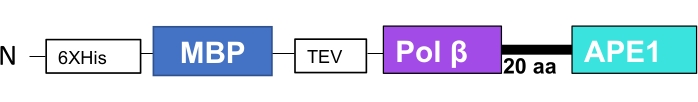
Figure 3: Schematic of genetically fused Polβ-APE1 with MBP at its N-terminal. MBP-Polβ-APE1 fusion protein was enzymatically characterized for both activities (data not shown). Please click here to view a larger version of this figure.
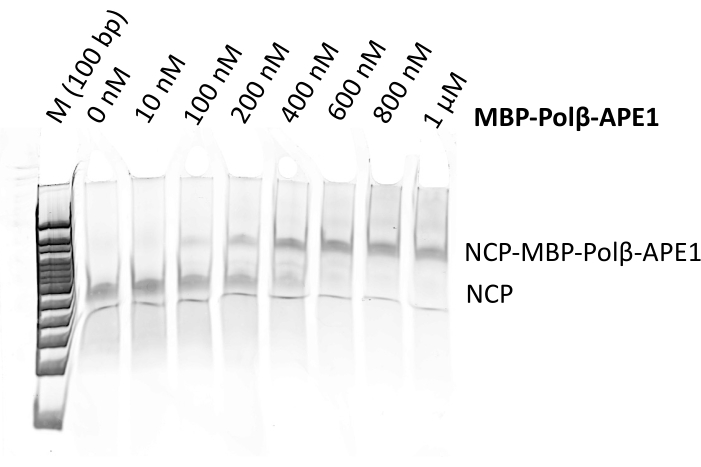
Figure 4: MBP-Polβ-APE1 binding to NCPs. Nucleosomal substrate (100 nM) was incubated with increasing amounts of MBP-Polβ-APE1 for 15 min on ice. Bound and unbound NCP were separated in a 6% polyacrylamide nondenaturing gel, followed by sybr green I staining. Please click here to view a larger version of this figure.
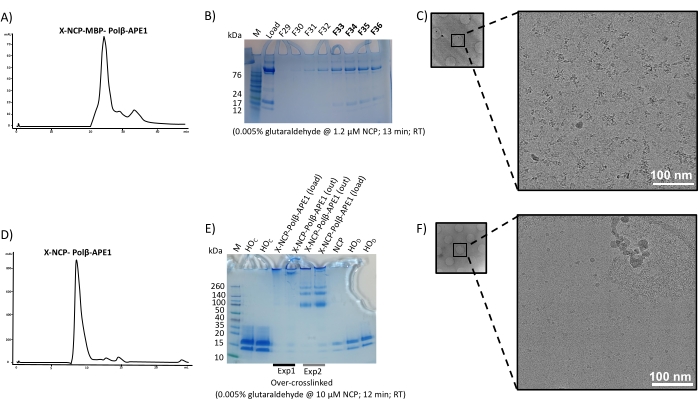
Figure 5: Analysis of successful and unsuccessful crosslinking results. The elution profile of the complex is shown in (A) with the peak of interest labeled. Fractions, denoted by "F", collected from this elution were analyzed in a 16% tricine polyacrylamide gel and shown in (B). (C) Sample from (B) was frozen using holey carbon support grids and imaged using the 200 kV field emission cryo-transmission electron microscope. Corresponding unsuccessful experiments are shown in (D), (E), and (F). Note these unsuccessful experiments contain the fusion protein without MBP. HOc and HOD denote histone octamer concentrated and diluted, respectively; RT denotes room temperature; "X" corresponds to crosslinking with glutaraldehyde. Scale bars = 100 nm (C, F). Please click here to view a larger version of this figure.
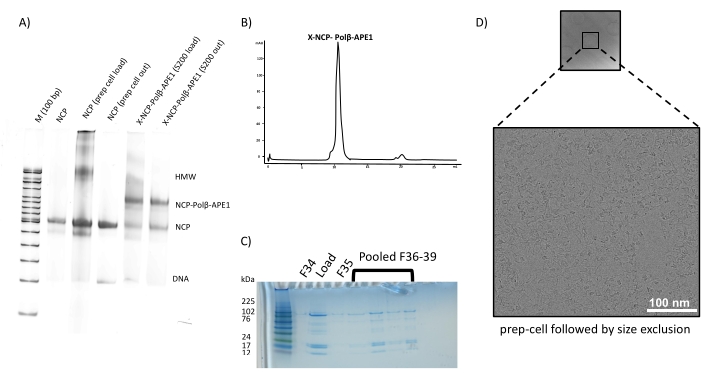
Figure 6: Preparation of NCP-Polβ-APE1 complex using the two methods in tandem. (A) Unbound and bound (1:1, NCP- Polβ-APE1) were analyzed in 6% polyacrylamide nondenaturing gel (note that at this ratio only 50% is bound). (B) Elution profile of NCP-Polβ-APE1 complex from a sizing column. (C) Analysis of eluted fractions in a 16% tricine polyacrylamide gel; fractions were pooled and concentrated, as indicated. (D) Sample was frozen and imaged as described in Figure 5B. Data processing did not show an additional density corresponding to Polβ-APE1 (not shown). "X" in lanes 5 and 6 denotes crosslinking with glutaraldehyde as indicated in the protocol. Scale bar = 100 nm (D). Please click here to view a larger version of this figure.
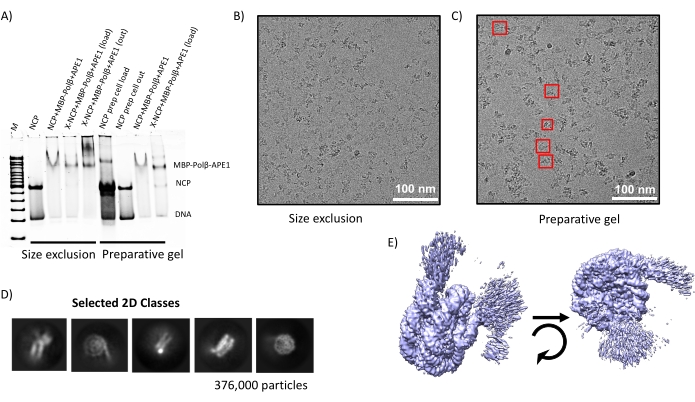
Figure 7: Analysis of NCP-MBP-Polβ-APE1 complex. (A) Samples from the different steps in the methods illustrated in Figure 1 were electrophoresed in a 6% polyacrylamide nondenaturing gel. Notice that complex formation is reproducible by the two methods. (B,C) Representative grid holey carbon support grid images taken using a 300 kV transmission electron microscope where the red boxes highlight different orientations of the complex (some of which correspond to the 2D classes shown in (D). (E) Data processed from (C) show a 3D map at 3.2 Å resolution of the NCP-MBP-Polβ-APE1 complex.). Scale bars = 100 nm (B, C). Please click here to view a larger version of this figure.
| Oligonucleotide name | Sequence (5'–>3') | Purification scale | Method of purificaiton | Number of vials ordered | ||
| UND complementary strand | TGATGGACCCTATACGCGGCCGCCCTGGAGAAT CCCGGTGCCGAGGCCGCTCAATTGGTCGTAGA CAGCTCTAGCACCGCTTAAACGCACGTACGCGC TGTCCCCCGCGTTTTAACCGCCAAGGGGATTAC TCCCTAGTCTCCAGGCACGTGTCAGATATCAAC ATCCTGTGCATGTATTGAACAGCGACCTTGCCG |
20 nmole | PAGE purified | 6 | ||
| 161mer | /5Phos/GATATCTGACACGTGCCTGGAGACTAGG GAGTAATCCCCTTGGCGGTTAAAACGCGGGGG ACAGCGCGTACGTGCGTTTAAGCGGTGCTAGA GCTGTCTACGACCAATTGAGCGGCCTCGGCA CCGGGATTCTCCAGGGCGGCCGCGTATAGGG TCCATCA |
20 nmole | PAGE purified | 4 | ||
| 35mer | CGGCAAGGTCGCTGTTCAATACATGCACAGG ATGT |
250 nmole | HPLC | 1 | ||
Table 1: DNA substrate. Three oligonucleotides were annealed to generate the dsDNA substrate. The underlined sections are 25 bp of linker DNA flanking the 147 bp 601 DNA positioning sequence containing a single nucleotide gap.
| Step | Temperature (°C) | Time (min)/step |
| 1 | 95 | 10 |
| 2-5 | 90, 85, 80, 75 | 5 |
| 6-41 | 69 decrease by 1 | 3 |
| 42 | 4 | hold |
Table 2: Annealing temperature gradient. Oligos were incubated in a thermocycler at these temperatures to generate the dsDNA substrate.
| Components (stock) | Volume/amount | Final concentration |
| 3M KCl | 167 mL | 0.250 M |
| 1 M HEPES, pH 8.0 | 20 mL | 10 mM |
| 0.5 M EDTA | 4 mL | 1 mM |
| 1 M DTT | 2 mL | 1 mM |
| CHAPS | 2 g | 1.6 mM |
| QS with dH2O to make 2L | ||
Table 3: RBlow reconstitution buffer. Instructions to make 2 L as previously reported14, except 1.6 mM CHAPS was added.
| Components (stock) | Volume/amount | Final concentration |
| 3M KCl | 267 mL | 2 M |
| 1 M HEPES, pH 8.0 | 4 mL | 10 mM |
| 0.5 M EDTA | 800 μL | 1 mM |
| 1 M DTT | 400 μL | 1 mM |
| CHAPS | 0.4 g | 1.6 mM |
| QS with dH2O to make 400 mL | ||
Table 4: RBhigh reconstitution buffer. Instructions to make 400 mL as previously reported14, except 1.6 mM CHAPS was added.
| Components (stock) | Volume | Final concentration |
| 3M KCl | 16.7 mL | 50 mM |
| 1 M HEPES, pH 8.0 | 10 mL | 10 mM |
| 0.5 M EDTA | 2 mL | 1 mM |
| 1 M DTT | 1 mL | 1 mM |
| QS with dH2O to make 1L | ||
Table 5: RB50mM reconstitution buffer. Instructions to make 1 L of reconstitution buffer compatible for freezing.
