Using this technique, the ramified and quiescent microglia can be chronically observed within all layers of the dorsal CA1, including stratum oriens (SO), stratum pyramidale (SP), stratum radiatum (SR), and stratum lacunosum-moleculare (SLM), especially 3-4 weeks after surgery when the inflammation subsides. If the surgery is performed appropriately, imaging can be performed up to several months after surgery. This section contains three topics useful for the evaluation of intravital imaging. First, sample images immediately following surgery and in the chronic phase are provided as references for appropriate surgical and imaging procedures. Second, the distinct morphology of microglia, their fluorescence intensity, and blood vessel distribution in specific hippocampal layers are described to help readers estimate the imaging depth ranging from the alveus to the DG. Third, an application example is provided that illustrates simultaneous imaging of neurons and microglia.
The representative images of microglia in CX3CR1-GFP mice immediately after surgery are shown in Figure 2. If there is no apparent damage to the CA1 and the curvature of the CA1 layers is properly flattened by the overlying coverslip, two-photon imaging depths of microglia exceed the entire CA1 layers and reach 500 µm from the surgical surface, where the ML or the granule cell layer (GCL) of the DG exist (Figure 2A; see step 4.7 and Figure 4E). Microglia are slightly less ramified, especially in the layer close to the surgical surface in comparison with those in the intact hippocampus. Nevertheless, they exhibit no prominent activation, suggesting proper surgical procedures (Figure 2B). On the other hand, edema in the CA1 induced by tissue damage will limit the imaging depth to 200 µm (Figure 2C). Tissue damage also induces abnormal microglial activation, such as accumulation of bulging processes toward the free tissue surface (Figure 2D; compare to the upper image of Figure 2B). These are signs of inappropriate surgery.
The representative images of microglia in the chronic phase more than 3 weeks after surgery are shown in Figure 3. Microglia can be imaged again beyond the CA1 to the deeper layers of the DG with higher fluorescent intensity and resolution and with more homogenous distribution (see step 6.3, Figure 3B). Imaging quality is better than that immediately after the surgery (Figure 2A). This difference may be explained by reduced edema and inflammation. Microglia in all layers have already restored their ramified morphology (Figure 3C), different from their postoperative appearance (Figure 2B). Time-lapse imaging of microglia in the CA1 reveals the immobile cell bodies and highly motile processes for surveillance (Video 1–2). Their motile behavior is similar to the previous report of microglial process dynamics in the cortex9.
It is straightforward to specify the imaged hippocampal layers based on the distribution and the depth of neuronal cell bodies, using mice expressing fluorescent probes in the hippocampal neurons25,26,27. Without the aid of the positional information about the neuron cell bodies, the signals from fluorescent microglia alone provide some clues to the identification of hippocampal layers (Figure 3B). Microglia in the alveus have elongated cell bodies and processes which tend to extend along the axonal tracts (Figure 4A). Microglia in other layers can be distinguished by their round cell bodies and processes radiating without preferred directions. Microglia in SP are among the densely packed pyramidal neurons and can be distinguished by the lower density of microglial processes. The layers above and below SP are identified as SO and SR, respectively (Figure 4B). It is impossible to distinguish between SR and SLM based on microglial properties alone. The border between the CA1 and the DG is recognized by thick blood vessels running along the boundary (Figure 4C). These vessels can be better recognized by labeling vessels with dyes such as sulforhodamine 101 (SR101; 5 mM, 4 µL/g body weight) injected intraperitoneally (Figure 4D). The fluorescence signal from microglia drops sharply in the DG compared to the overlying CA1, probably due to the difference in the refractive index of these structures. This gap in fluorescence intensity may also help to specify the border between the DG and CA1. As an application example, simultaneous imaging of GFP-positive microglia and tdTomato-labeled pyramidal neurons is shown (Figure 4E). In this experiment, CX3CR1-GFP mice received an injection of adeno-associated virus (AAV) for the expression of tdTomato in the hippocampal pyramidal neurons. Neuronal labeling will help to understand the exact layer structures of the CA1.
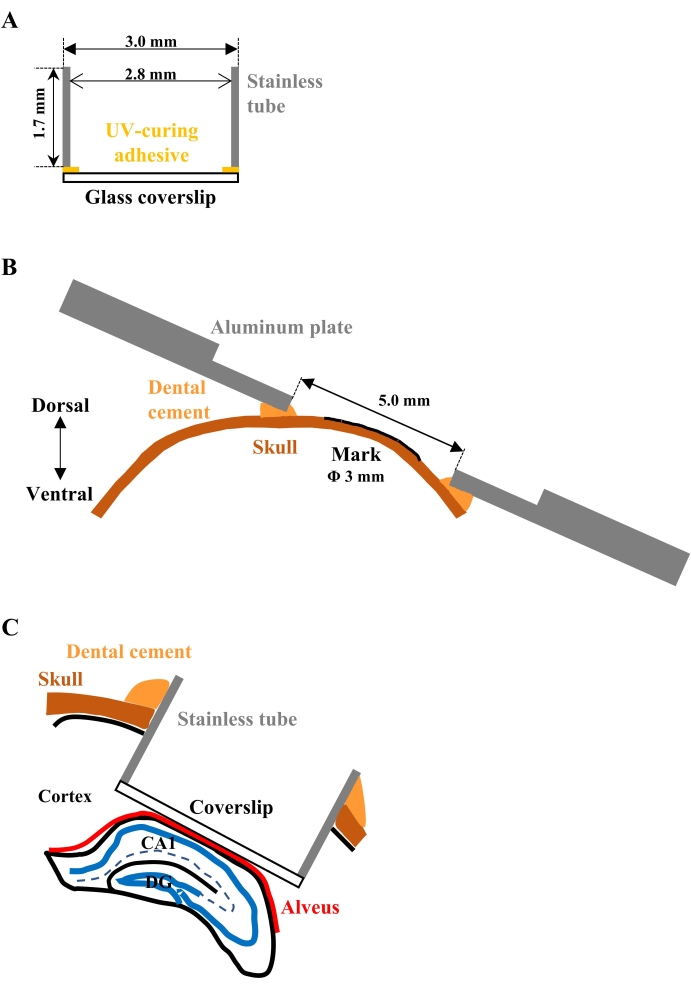
Figure 1: Schematic drawings for the implantation of the observation window. (A) Cross-sectional view of the assembled glass-bottom metal tube. A circular glass coverslip adheres to the one end of the cylindrical stainless tube. (B) Cross-sectional view of the aluminum plate attached to the skull. The plate should be parallel to the marked surface of the skull so as not to interfere with the objective lens, positioned close to the plate for focusing. (C) Cross-sectional view of the implanted tube. The glass bottom should be closely attached to the alveus of the hippocampus to gently press and flatten the surface of the CA1. Please click here to view a larger version of this figure.
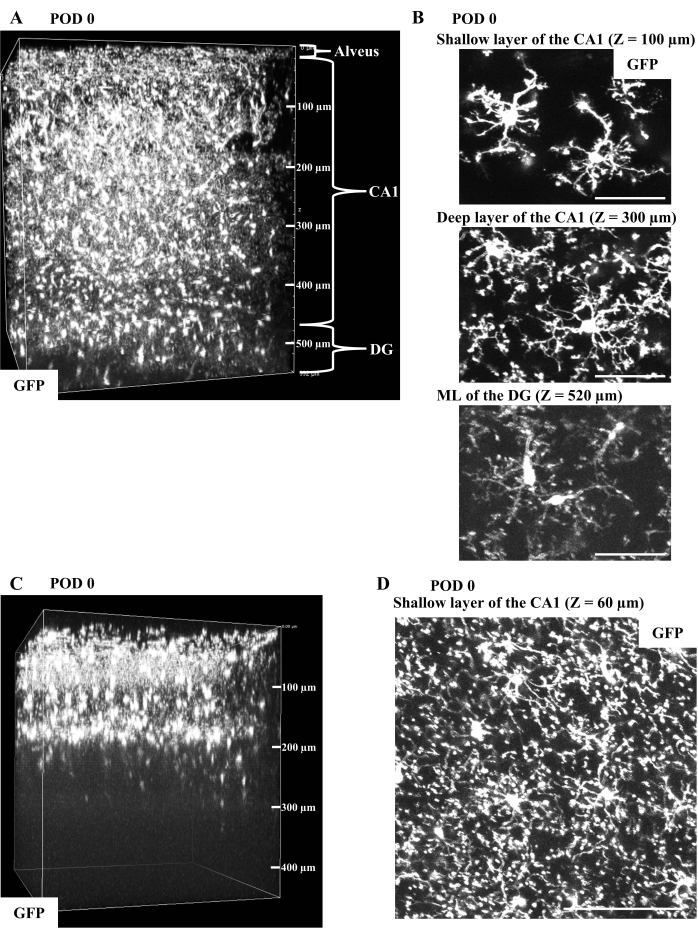
Figure 2: In vivo imaging of microglia in CX3CR1-GFP mice immediately after surgery. (A) Representative 3D reconstruction from the in vivo CA1 imaging of a CX3CR1-GFP mouse on postoperative day (POD) 0 (width: 511 µm, height: 511 µm, depth: 550 µm). The fluorescent microglia in the ML of the DG at a depth of 500 µm from the glass bottom can be imaged through the entire CA1. (B) Typical GFP-filled microglia obtained in (A) are shown as the maximum intensity projections (MIPs) of image stacks with 20 µm thickness at depths of 100, 300, and 520 µm from the glass bottom. Microglial morphology is detectable from the superficial CA1 to the ML of the DG, though with apparent image deterioration in the DG. Microglia especially in the layer close to the surgical surface are less ramified but exhibit no obvious activation. Scale bars, 50 µm. (C) Representative 3D image reconstruction of the surgically damaged CA1. The data was acquired from a CX3CR1-GFP mouse on POD 0 (width: 399 µm, height: 399 µm, depth: 450 µm). Microglial fluorescence is detectable only up to the depth of 200 µm, in contrast with the undamaged CA1 (A) with fluorescent microglia detected at a depth of 500 µm. (D) The typical image of abnormally activated microglia in (C). The MIP of GFP image stacks with 20 µm thickness at a depth of 60 µm shows microglial bulging processes extending toward the surgically exposed tissue surface. The overall microglial morphology is different from the image shown in (B). Scale bar, 100 µm. Please click here to view a larger version of this figure.

Figure 3: In vivo imaging of microglia in CX3CR1-GFP mice in the chronic phase. (A) The representative appearance of the implanted window on the right dorsal CA1 in the chronic phase. Through the glass bottom, the white fibers of the alveus are clearly observed without postoperative bleeding. The DHC and the LV are partly visible in the caudomedial area and in the rostrolateral edge, respectively. Scale bar, 1 mm. (B) Representative 3D reconstruction from the in vivo chronic CA1 imaging of a CX3CR1-GFP mouse on POD 24 (width: 511 µm, height: 511 µm, depth: 670 µm). Compared to the images immediately after surgery (Figure 2A), microglia show a more homogeneous distribution and can be observed more clearly in the deeper layers of the DG because of the reduction of edema and inflammation. (C) Typical images of microglia from (B) with fully restored ramified morphology are shown as the MIPs of GFP image stacks with 20 µm thickness at depths of 100, 280, and 500 µm from the surgical surface. Scale bars, 50 µm. Please click here to view a larger version of this figure.
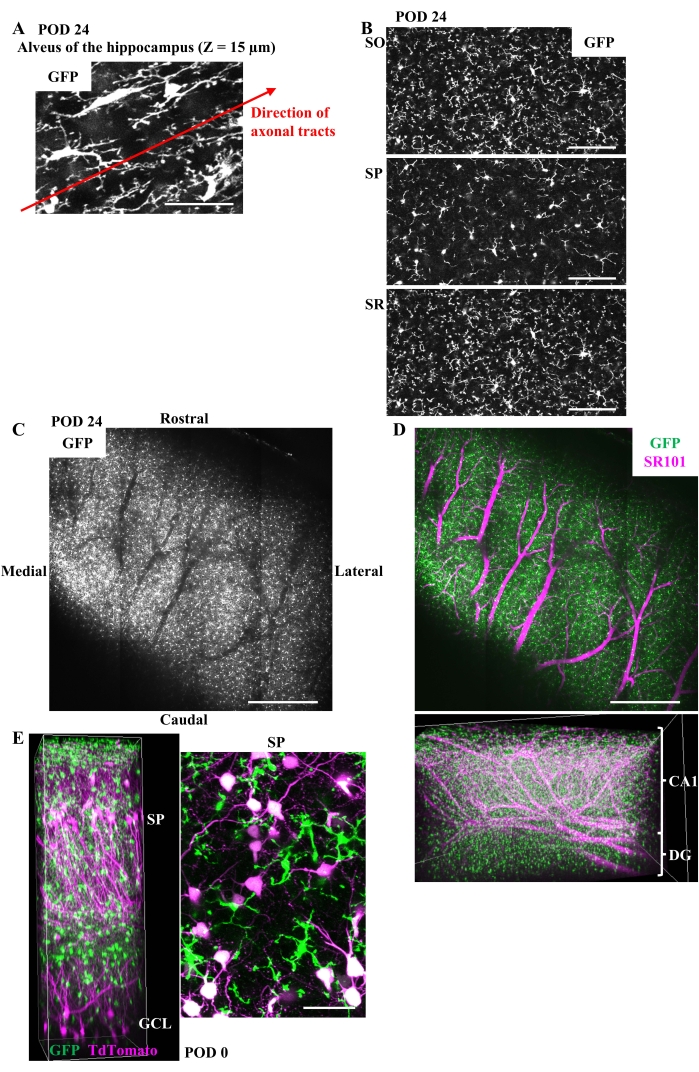
Figure 4: Clues to identifying each layer of the hippocampus during in vivo imaging and an application example of this method. (A) The typical image of microglia in the alveus of CX3CR1-GFP mice in the chronic phase. Microglial cell bodies and processes extending along the axonal tracts are shown in the MIP of GFP image stacks with 10 µm thickness at a depth of 15 µm. Scale bar, 50 µm. (B) Representative images of microglia in SO, SP, and SR in the chronic phase shown as the MIP of GFP image stacks with 5 µm thickness. The local density of microglial processes is lower in SP than in surrounding SO or SR due to the densely packed pyramidal neurons in SP. Scale bars, 100 µm. (C) The thick blood vessels running along the boundary between the CA1 and the DG can be recognized only by the microglial fluorescence. The average intensity projection of tiled GFP image stacks with 90 µm thickness in the chronic phase at a depth of about 500 µm is shown. The void of the GFP signal corresponds to the thick vessels. Scale bar, 500 µm. (D) (Upper) The image of the same field of view as C after intraperitoneal injection of SR101. Scale bar, 500 µm. (Lower) 3D reconstruction of the tiling images after SR101 injection (width: 2.60 mm, height: 1.73 mm, depth: 0.69 mm). The thick vessels running along the border between the CA1 and the DG can be easily recognized by the intravascular SR101 fluorescence. (E) (Left) 3D reconstruction of the CA1 and the DG (width: 255 µm, height: 255 µm, depth: 730 µm) and (right) the MIP of GFP and tdTomato fluorescence in SP made from image stacks with 20 µm thickness. At 1 week before surgery, 1.5 µL of a mixture of AAV1-CAG-FLEX-tdTomato (5.0 x 1012 vg/mL) and AAV1-hSyn-Cre (1.0 x 109 vg/mL) was injected into the hippocampus of a CX3CR1-GFP mouse to label the neurons sparsely. Imaging was performed on POD 0. SP and GCL are easily recognized by the position of neuronal cell bodies. Scale bar, 50 µm. Please click here to view a larger version of this figure.
Video 1: Time-lapse imaging of SO microglia in the chronic phase. The MIP of GFP image stacks with 20 µm thickness in the time-lapse imaging of SO microglia, animated so that 28 min play in 1 sec. Scale bar, 50 µm. Please click here to download this Video.
Video 2: Time-lapse imaging of SR microglia in the chronic phase. The MIP of GFP image stacks with 20 µm thickness in the time-lapse imaging of SR microglia, animated so that 28 min play in 1 sec. Scale bar, 50 µm. Please click here to download this Video.
