The esophagus is divided into different layers: epithelium, lamina propria, submucosa, and muscularis externa (Figure 1A). Fibroblasts reside within the submucosa and lamina propria, referred to as the stroma. In this protocol, the muscularis externa is mechanically removed (Figure 1B), which does not lead to a loss of fibroblasts (PdgfrαH2BeGFP+) residing in the stroma (Figure 1C). Before dissociation, the epithelium is separated from the stroma resulting in two tissue segments (Figure 1B). Separating the two layers provides the opportunity to increase dissociation time for the more robust epithelial layer compared to the fragile stromal layer. In this way, an efficient isolation protocol yielding both viable epithelial progenitor cells as well as stromal fibroblasts is established (Figure 1B). Esophageal progenitor cells are sorted based on their high INTEGRIN-β4 and E-CADHERIN expression (Figure 1C,D).
Subpopulations of fibroblasts can be isolated by using distinct markers. In this protocol, a strategy for fibroblast isolation based on commonly used fibroblast markers PDGFRα and DPP4 (CD26) is provided. Isolation by either the PdgfrαH2BeGFP reporter expression or DPP4 antibody shows that around 50% of the isolated cells are fibroblasts (Figure 1E,F). Additionally, 70% of the PDGFRα+ fibroblasts are DPP4+, indicating that a largely overlapping, but not identical, fibroblast population is obtained. After isolating both epithelial and stromal cell populations, esophageal progenitor cells are either cultured alone or together with fibroblasts in a matrix dome. To study the contribution of fibroblasts to organoid formation, the co-culture is maintained in a growth factor reduced medium (Figure 1G).
Esophageal progenitor cells form organoids in the presence of EGF, NOGGIN and RSPO (ENR). Removing NOGGIN and reducing the amount of RSPO (25 ng/µL; ERlow) is sufficient to prevent organoid formation (Figure 2A). Interestingly, adding either DPP4+ or PDGFRα+ fibroblasts to the esophageal progenitor cells in the ERlow medium restores the organoid forming ability, demonstrating a supportive function for both fibroblast populations (Figure 2A–D). Visualization of the PdgfrαH2BeGFP transgene shows that fibroblasts are in close contact with the epithelial progenitor cells during organoid formation (Figure 2A). At day 6, PdgfrαH2BeGFP+ fibroblasts are still abundantly present in the co-culture. Fibroblasts are present throughout the dome, near and touching the organoids (full arrow), or attached to the organoids (arrowhead; Figure 2D).
Whole mount staining shows a 3D representation of the interaction of the fibroblasts with the organoids (Figure 3). While it is possible to perform the whole mount protocol without the use of a clearing solution, it decreases transparency and laser penetration of the organoid (Figure 3B, z-view). When mounting organoids, the spacer helps to maintain organoid morphology. In contrast, plating the coverslip directly on the organoids (without a spacer) flattens the organoids and results in loss of organoid structure (Figure 3A,B).
Both the DPP4+ and PDGFRα+ fibroblasts are found to be wrapped around the organoids (Figure 3C, Video1, and Video 2). Differentiation of esophageal organoids can be assessed using different markers. Figure 4 shows that the staining protocol provided is suitable for easier-to-stain keratins (KRT14/13) as well as more-difficult-to-stain transcription factors (TRP63/KLF4). The co-culture protocol generates organoids with a similar differentiation pattern, as demonstrated in vivo13,14 and as seen in organoids grown in ENR medium; KRT14+ or TRP63+ progenitor cells form the outer layer and KRT13+ or KLF4+ differentiated cells oriente inwards.
This protocol provides a tool to study the esophageal stem cell niche in vitro and visualizes the interaction between organoids and fibroblasts. By implementing a protocol for the isolation of fibroblasts using antibodies, the method is adaptable and can be used to study fibroblast subpopulations without the need of transgenic mice.
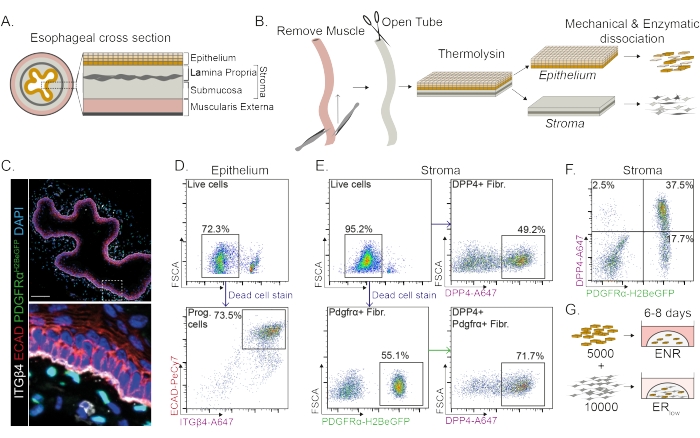
Figure 1: Isolation of progenitor cells and fibroblast subpopulations from the esophagus. (A) Schematic overview of the different layers in the esophagus. The stroma contains the lamina propria and submucosa. (B) Schematic overview of the isolation protocol. The muscle (muscularis externa) is mechanically removed using forceps; the remaining esophagus is cut open and incubated in thermolysin to separate the epithelial layer from the stroma. The epithelium and stroma are separated, mechanically minced, and enzymatically digested to single cell suspensions. Dissociated cells are then stained and prepared for FACS. (C) Cross section of the esophagus stripped from the muscularis externa showing PdgfrαH2BeGFP+ fibroblasts in the stroma. INTEGRIN-β4 (ITGβ4) and E-CADHERIN (ECAD) double positive cells are the epithelial progenitor cells of the esophagus. Scale bar = 100 µm. (D) Representative flow cytometry plot of epithelial cell isolation showing the percentage of live cells (upper panel) from all single cells. The lower panel shows the percentage of isolated ITGβ4+ ECAD+ progenitor (Prog.) cells from all live cells. (E) Representative flow cytometry plot of stromal cell isolation showing the percentage of live cells (upper left panel). Representative flow cytometry plots showing the percentage of isolated DPP4+ fibroblasts (Fibr.; upper right panel) and Pdgfrα+ fibroblasts (lower left panel) of all live cells. 70% of the Pdgfrα+ fibroblasts are also DPP4+ (lower right panel). (F) Representative flow cytometry plot of the stroma showing DPP4+ only cells (2.5%), DPP4+ PDGFRα+ cells (37.5%), and PDGFRα+ only cells (17.7%). The percentages are of all live cells. (G) Epithelial only cells are plated in a matrix dome in the presence of 50 ng/µL EGF, 100 ng/µL NOGGIN, and 250 ng/µL RSPO (ENR), or together with fibroblasts in the presence of EGF and a low concentration of RSPO (25 ng/µL). Please click here to view a larger version of this figure.
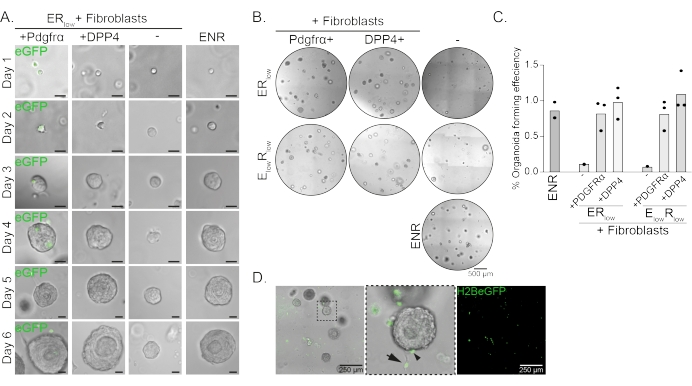
Figure 2: Representative results of organoid co-cultures. (A) Brightfield images showing growth of the organoids from day 1 to day 6. The brightfield images with the organoids co-cultured with PdgfrαH2BeGFP+ fibroblasts also show the nuclear eGFP signal. Scale bar = 25 µm. (B) Brightfield images of the whole matrix dome at day 6. The left column shows organoid co-cultures grown in the presence of Pdgfrα+ fibroblasts in ERlow or ElowRlow medium. The middle column shows organoid co-cultures grown in the presence of DPP4+ fibroblasts in ERlow or ElowRlow medium. The right column shows organoid mono-cultures grown in ENR medium. ENR medium = EGF (50 ng/µL), NOGGIN (100 ng/µL), and RSPO (250 ng/µL). ERlow = EGF and 25 ng/µL RSPO. ElowRlow = 5 ng/µL EGF and 25 ng/µL RSPO. Scale bar = 500 µm. (C) Graph showing the organoid forming efficiency (%) (i.e., the percentage of cells forming organoids in different culture conditions). Each dot represents a matrix dome and the bar represents the mean of all dots per condition. (D) Brightfield and fluorescent image of day 6 organoids co-cultured with PdgfrαH2BeGFP+ fibroblasts. PdgfrαH2BeGFP+ fibroblasts are present throughout the dome, attached to the organoids (arrowhead), and unattached but in contact with the organoids (full arrow). Scale bar = 250 µm. Please click here to view a larger version of this figure.
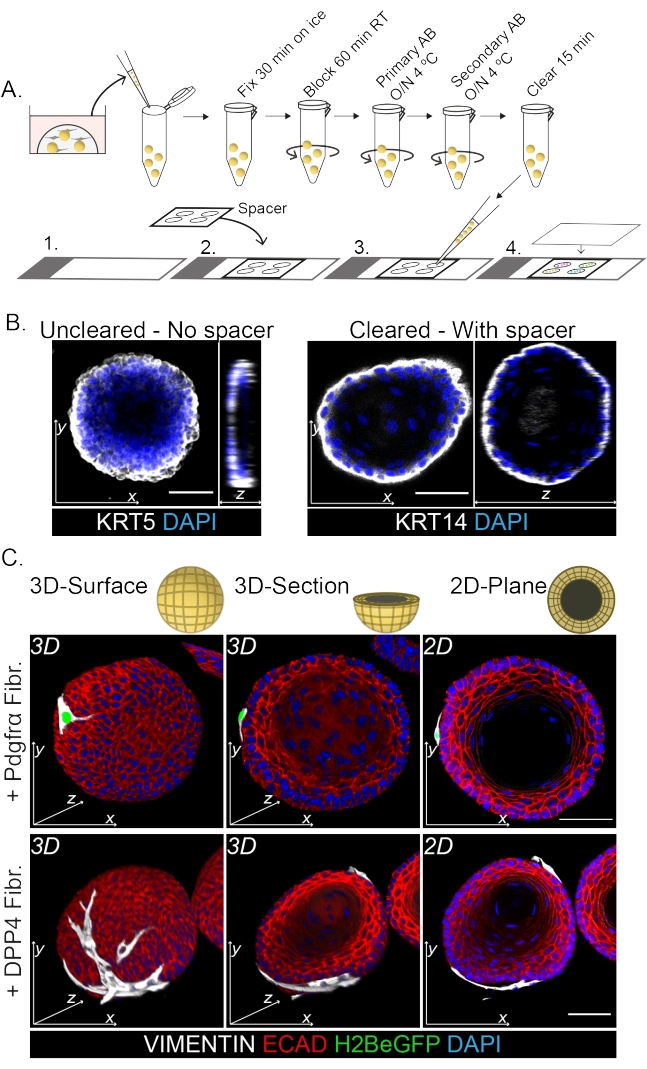
Figure 3: Whole mount staining protocol for the study of fibroblast-organoid interactions. (A) Schematic overview of the whole mount immunofluorescence protocol. AB = antibody. (B) Immunofluorescence picture of uncleared whole mount staining showing a decreased transparency and penetration of the laser light compared to the cleared organoids. The absence of a spacer results in flattening of the organoid and loss in organoid morphology. (C) Whole mount images of the co-cultured organoids show 3D surfaces of the organoids with VIMENTIN+ fibroblasts (Fibr.) wrapped around and in close contact with the organoid. 3D cross sections and 2D plane images show the lumen of the organoid. Scale bar = 50 µm. Please click here to view a larger version of this figure.
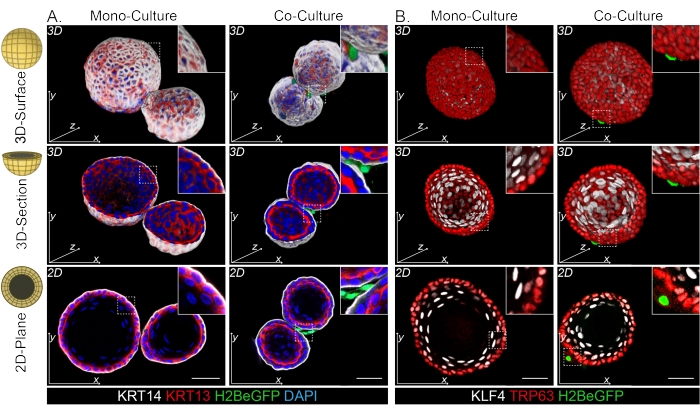
Figure 4: Whole mount images reveal distinct basal and suprabasal cell populations. (A) Whole mount staining of mono- and co-cultured organoids with PdgfrαH2BeGFP+ fibroblasts showing KRT14+ basal cells in the outer layer and KRT13+ differentiated suprabasal cells. Scale bar = 50 μm. (B) Whole mount staining of mono- and co-cultured organoids with PdgfrαH2BeGFP+ fibroblasts showing TRP63+ basal cells in the outer layer and KLF4+ differentiated suprabasal cells. Scale bar = 50 µm. Please click here to view a larger version of this figure.
Table 1: Table describing the organoid culture media components. Please click here to download this Table.
Video 1: PdgfrαH2BeGFP+ fibroblast wrapped around and in close contact with the organoid. The video accompanies the upper panel of Figure 3C. The scale bar in Figure 3C is 50 µm, and the organoid is ~120 µm in diameter. VIMENTIN is shown in white, E-CADHERIN in red, PdgfrαH2BeGFP in green, and DAPI in blue. Please click here to download this Video.
Video 2: DPP4+ fibroblast wrapped around and in close contact with the organoid. The video accompanies the lower panel of Figure 3C. The scale bar in Figure 3C is 50 µm, and the organoid is ~120 µm in diameter. VIMENTIN is shown in white, E-CADHERIN in red, and DAPI in blue. Please click here to download this Video.
