The protocol described in this study is based on the fluorescence imaging and electrical stimulation studies conducted by Weitz et al.12. The protocol consists of three main parts: (1) fluorescent labeling of the GCL and flat-mounting of the retina on the MEA (Figure 1-left), (2) visualization of calcium activity in the GCL during electrical stimulation (Figure 1-middle), and (3) extraction, processing, and interpretation of the imaging data (Figure 1-right).
First, as depicted in Figure 1-left, Long Evans rats are intravitreally injected with AAV2-CAG-GCaMP5G prior to the imaging session. The optimal viral expression for this vector occurs 2 to 3 weeks after injection12,18. After fully anesthetizing the animal, a pilot hole is made using a 30 G needle, and then 5 µL of AAV2-CAG-GCaMP5G is slowly injected into the vitreous using a 36 G blunt needle attached to a precision syringe to prevent reflux. During viral expression, an in vivo retinal imaging system is used to assess the condition of the retina post-surgery, with OCT images providing detailed visualization of the retinal layers. Once gene expression is achieved, the retina is carefully extracted from the eyecup using a stereo microscope and high-precision dissection tools. From this point onwards, the tissue is manipulated in oxygenated media to preserve the sample. The excised retina, with the GCL facing up, is then mounted on a platform designed for flat-mounting to ensure stability and prevent sample floating. The sample is mounted on the MEA surface with the GCL facing the electrodes.
Next, the MEA is mounted on its interface board on an inverted fluorescent microscope (Figure 1-middle). The retinal sample is perfused with oxygenated media at 33 °C using a perfusion system. The sample can be maintained in this configuration for several hours. The desired stimulation scheme is programmed, and images are acquired at a rate of 10 frames per second. It is recommended to name the movies according to the applied electrical stimulation parameters. Image acquisition should begin before the initiation of stimulation to obtain some baseline frames without stimulation, which will serve as a negative control.
Finally, as illustrated in Figure 1-right, the data is extracted from the time-lapse images by segmenting the cell somas. Photobleaching effects are corrected by fitting the data, and responsive cells are identified. Responsive cells are defined as those with fluorescence peaks during stimulation that exceeds their baseline by 2.5 times. If a cell responds to three out of the five bursts of stimulation, it is considered responsive to that specific train of stimulation.
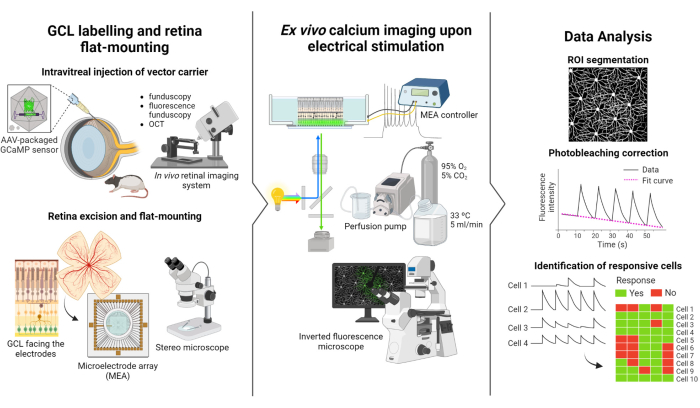
Figure 1: Overview of the study. Schematic illustration of the protocol for (left) fluorescently labeling the GCL of the retina and sample mounting, (middle) set up preparation for ex vivo recordings with electrical stimulation provided by a MEA, and (right) analyzing the calcium imaging data to classify responsive cells. Please click here to view a larger version of this figure.
Intravitreal-injected retina
The incidence of complications associated with intravitreal injections is very low. However, there are some complications that can arise from the surgery itself, regardless of the injected component. These complications include cataract formation, vitreous hemorrhage, elevation of intraocular pressure, and endophthalmitis23. To determine whether these complications are caused by the surgery, the animal needs to undergo evaluation before the procedure using funduscopy and OCT. Three days after the injection, the animals should be followed up. In Figure 2A–D, the retina of a healthy injected animal is shown. After two weeks of injection, the RGCs begin to express fluorescence, which can be visualized using fluorescence fundoscopy (Figure 2B,C). OCT images provide detailed visualization of the disposition and thickness of retinal layers (Figure 2D), offering higher resolution compared to funduscopy, particularly when assessing retinal detachment. Once the retina is flat-mounted and imaged using an inverted fluorescence microscope, it becomes possible to distinguish the cells and axon bundles. Unlike other calcium indicators, the GCaMP indicator is restricted to the cytoplasm7, and fluorescence is excluded from the nucleus (Figure 2E).
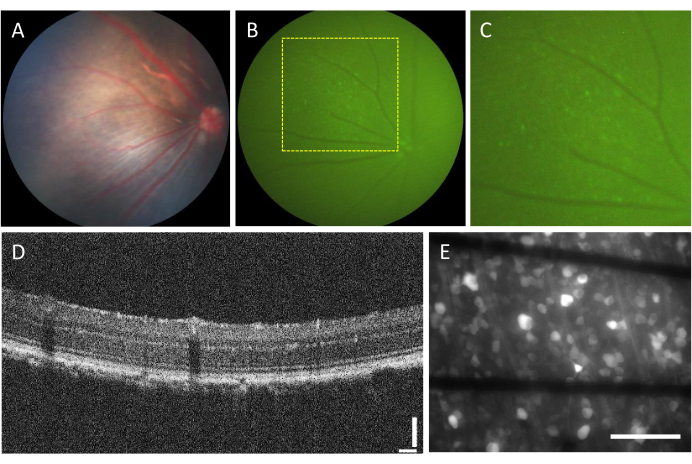
Figure 2: Representative images of the intravitreal-injected retina. (A) Fundoscopy, (B) fluorescence fundoscopy, (C) zoom-in of the fluorescence fundoscopy, (D) OCT image, and (E) epi-fluorescence image of the excised retina mounted on a custom MEA with graphene-based electrodes on 500 µm thick borosilicate glass. In (E), black lines correspond to Ti/Au traces. Scale bars: 115 µm (D) and 100 µm (E). Please click here to view a larger version of this figure.
Electrodes and GCL contact
In order to evoke neural responses effectively, it is crucial to ensure that the flat-mounted retina is in close contact with the surface of the MEA. A simple way to verify this is by visually confirming whether the cells and electrodes are located in the same focal plane (Figure 3A). If the cells are not in the same focal plane as the electrodes (Figure 3B), it indicates that the contact is suboptimal, which will result in less effective stimulation.
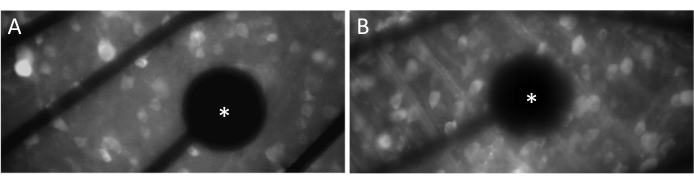
Figure 3: Electrodes and GCL contact. (A) Cells and the electrode (asterisk) in the same focal plane. (B) Cells and electrodes not in the same focal plane, indicating suboptimal contact for electrical stimulation in that area. Please click here to view a larger version of this figure.
Ex vivo calcium imaging upon electrical stimulation provided by a MEA
The resulting data from calcium imaging consist of time-lapse images that monitor the neural activity of hundreds of cells in response to electrical stimulation. Suprathreshold stimuli cause a calcium influx into the cell somas, resulting in a sudden change in fluorescence intensity (Video 1). This protocol enables determining whether an electrode, MEA, and/or stimulation algorithm elicits the desired response in neural tissue. The size and pitch of the electrodes on the MEA, as well as the proportion of tissue being studied, will determine the appropriate objective magnification to choose. Typically, for single-electrode stimulation studies with diameters ranging from 5 µm to 100 µm, a 20-25x objective magnification is suitable (Figure 4A), providing a FOV of approximately 600 µm x 600 µm. For experiments involving stimulation with multiple electrodes, a 4-10x objective magnification may be necessary to assess a wider area of around 2 mm x 2 mm. Responsive cells can be easily identified by generating a standard deviation image projection of the time-lapse movie (Figure 4B and Video 1).
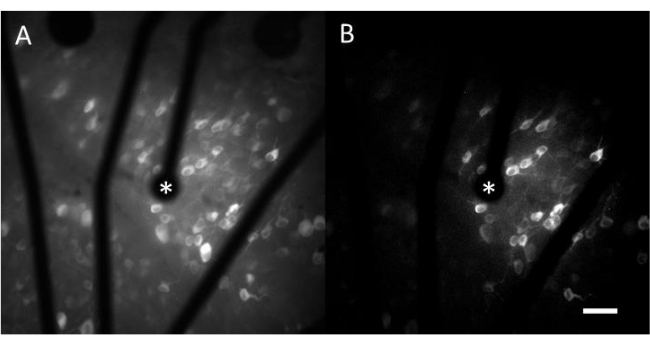
Figure 4: Calcium imaging of the GCL with electrical stimulation provided by a 25 µm-diameter electrode. (A) Maximum projection of a 60 s time-lapse movie and (B) standard deviation projection clearly depicting cells that respond to electrical stimuli from a 25 µm diameter porous graphene-based electrode. The stimulating electrode is indicated with an asterisk. Scale bar: 50 µm. Please click here to view a larger version of this figure.
Analysis of the calcium dynamics over time upon controlled stimulation
For each identified cell soma, the mean intensity values were extracted over time. Figure 5A shows the photobleaching-corrected calcium traces from the responsive cells. In this example, five bursts of biphasic pulse trains (cathodic-first, 40 cycles, 1 ms duration, 2 µA amplitude) were delivered every 10 s (indicated by black lines) during a 60 s image acquisition. Within a given experiment, the same five pulse trains are applied to test the consistency of the response. The frames captured during the non-stimulating periods (highlighted in red) are used to perform a linear fit, correcting for the photobleaching effect.
Once the responding cells are identified, and their coordinates (x,y) are known relative to the stimulating electrode, one can examine the relationship between the current required to activate the cells and the distance from the stimulating electrode (Figure 5B). As expected, cells located closer to the stimulating electrode require lower current values to evoke a response.
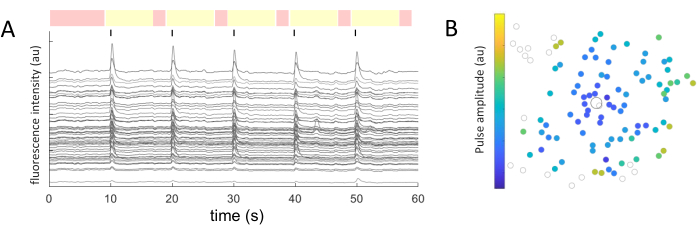
Figure 5: Representation of the electrical-evoked responses. (A) Calcium traces of cell somas upon 5 bursts of pulse trains (biphasic, cathodic-first, 40 cycles, 1 ms duration, 2 µA amplitude) every 10 s (black lines) during a 60 s image acquisition. Non-stimulating (red-highlighted frames) and stimulating periods (yellow-highlighted frames) are shown. Traces surpassing the baseline signal (root mean square of the non-stimulating periods) by 2.5 times are considered evoked responses. Cells responding in three out of the five stimulating periods are classified as responding cells. (B) Calcium activity distribution map showing the stimulating electrode (black outlined circle) and cells (gray outlined circle). The color code represents the minimum pulse amplitude necessary to evoke a cellular response. Please click here to view a larger version of this figure.
Video 1: Calcium imaging of the GCL with electrical stimulation provided by a 25 µm-diameter electrode. The video displays differences in fluorescence intensity due to electrical stimulation from a 25 µm diameter porous graphene-based electrode. The left side shows the original movie, and the right side shows the standard deviation projection where responding cells can be easily identified. Scale bar: 50 µm. Please click here to download this Video.
