Four patients with GBM and one with LGG were included after pathological confirmation by an experienced neuropathologist (CMM). The majority of patients had an unmethylated MGMT promoter, and all GBM patients were IDH1 and IDH2 wild type (Table 1). On average, the manufacturing process lasted 88.8 min (+/- 6.3 min) in the C approach and 322 min (+/- 17.2 min) in the M approach. The overall success rate was 87% in the manual and 93% in the chopper approach after 4 weeks of culture (n = 5). Moreover, PDOs derived from the C group reached the desired rounded shape within 1 week and were mature enough to be used in in vitro experiments, while the PDOs of the M group mostly remained sharply edged and undefined (Figure 2). The tumor tissue processed with the C approach resulted in overall 281 PDOs (Mean per patient = 56 +/- 43) after the first week of culture, while 250 PDOs (Mean per patient= 50 +/- 41) developed with the M approach. During the second week of culture, the tissue of all five patients yielded higher PDO numbers when they were generated with the C approach (801; Mean per patient= 130 +/- 38) compared to the M approach (601; Mean per patient= 76 +/- 44; P = 0.018). During the third week of culture, the C approach accumulated overall 1105 PDOs from all patients (Mean per patient = 221 +/- 32) compared to 771 PDOs (Mean per patient= 155 +/- 34) in the M approach (P = 0.032). Furthermore, a total of 1195 PDOs (Mean per patient= 239 +/- 50) formed after four weeks of culture when generated with the C approach compared to 784 (Mean per patient= 157 +/- 36) utilizing the M approach (P < 0.01). Therefore, the C method showed a significantly higher PDOs count starting from the second week (Figure 3). Furthermore, the relative fluctuations in PDO counts were evaluated to explore the dynamic trends between successive weeks. The analysis unveiled an impressive surge in PDO counts during the initial transition from the first to the second week in the C approach (265%), which was indicative of rapid progress. Subsequently, there was a lower rise in counts during the third week (75%), reflecting a temporary adjustment. In contrast, the M approach demonstrated a consistent and steady increase in PDO counts (92% in second week, respectively 67% in the third week), which contributed to a remarkable stability in counts during the fourth week. This consistent upward trend in PDO counts underscores the reliability and resilience of the C approach throughout the observation period.
Two GBM patients and one LGG patient were included for the analysis of astrocyte-numbers (GFAP) within PDOs, PDO cell proliferation (Ki67), and apoptosis (CC3). The determined astrocyte count revealed no significant differences between the two processing methods with an average of 43% in the C approach and 45% in the M approach (Figure 4 and Figure 5, Supplementary Figure 1 and Supplementary Figure 2). Similarly, the proliferation rates within the PDOs were comparable between the C (3%) and M approaches (1%). Only PDOs generated with the C approach from patient 5 displayed a proliferation rate of 26% compared to 1% with the M approach (P = 0.001; Figure 4C). Overall low apoptosis rates were detected in PDOs processed with the C approach (3%) compared to 2% from the M approach for all patients, which were not significantly different (Figure 5C). Furthermore, there was no significant difference between the two methods regarding the number of astrocytes undergoing apoptosis (Figure 5D).
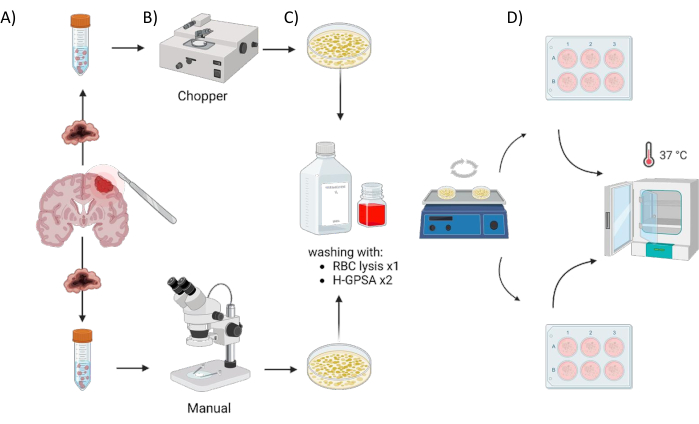
Figure 1: Graphical overview of the patient-derived organoid (PDO) manufacturing process using an automated chopper versus a manual approach. The illustration depicts the various steps involved, including (A) sample collection, (B) tumor material dissection, (C) washing, and (D) incubation. Please click here to view a larger version of this figure.
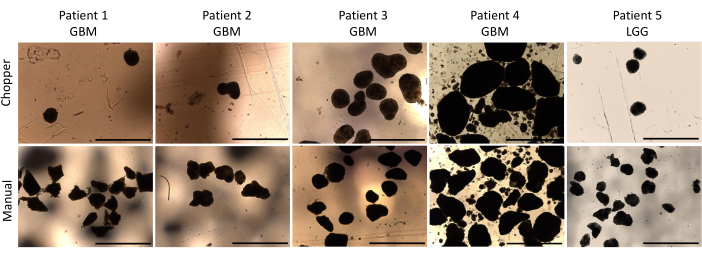
Figure 2: PDO morphology during the first week of culture. Comparison of PDOs formation post-dissection using both the automated chopper and manual method. Scale bars = 1000 µm. Please click here to view a larger version of this figure.
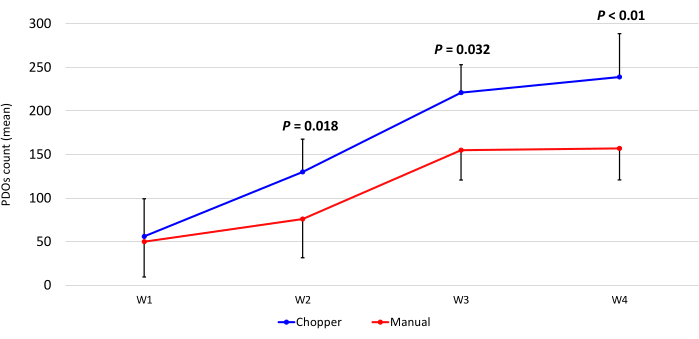
Figure 3: PDO count during the first four weeks of culture. The x-axis displays the time in weeks (W), and the y-axis the number of PDOs of the C (blue) and M (red) approach (n = 5). Each data point represents the mean count, with error bars indicating the standard error of the mean. Please click here to view a larger version of this figure.
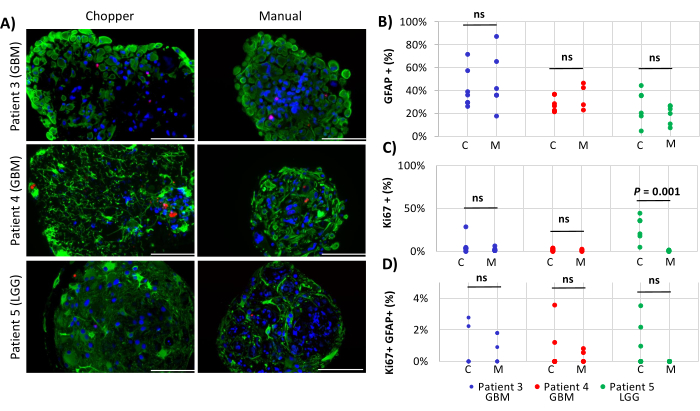
Figure 4: Cell proliferation rates within PDOs. (A) Representative immunofluorescence images (n = 3) of GFAP-positive cells (green), Ki67-positive cells (red), and DAPI (blue) in PDOs from patients 3, 4 and 5 (Table 1). All PDOs were processed using the chopper (C) and manual (M) methods. Scale bars = 100 µm. (B) Comparison of the two methods regarding the relative number of GFAP-positive cells, (C) Ki67-positive cells, and (D) Ki67/GFAP-double positive cells. Not significant results are indicated by "ns". Please click here to view a larger version of this figure.
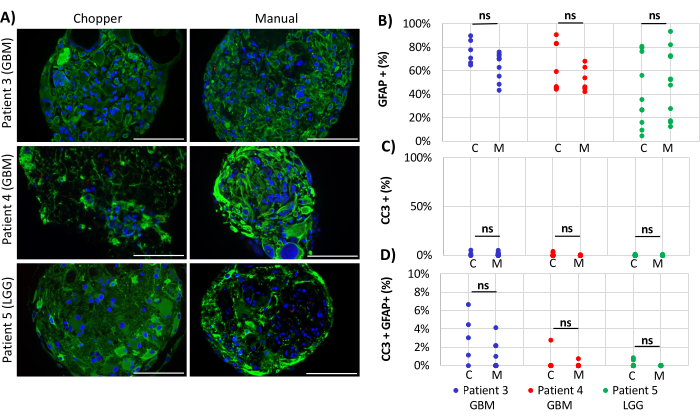
Figure 5: Apoptosis rates within PDOs. (A) Representative immunofluorescence images (n = 3) of GFAP-positive cells (green), CC3-positive cells (red), and DAPI (blue) in PDOs from patients 3, 4 and 5 (Table 1). All PDOs were processed using the chopper (C) and manual (M) methods. Scale bars = 100 µm. (B) Comparison of the two methods regarding the relative number of GFAP-positive cells, (C) CC3-positive cells, and (D) CC3/GFAP-double positive cells. Not significant results are indicated by "ns". Please click here to view a larger version of this figure.
Table 1: Patients' characteristics and clinical parameters. GBM = glioblastoma, IDH-wild type, CNS WHO grade 4; LGG = low grade glioma; KPS = Karnofsky performance score; MGMT = O6-methylguanine-DNA methyltransferase; IDH1 = isocitrate dehydrogenase 1, IDH2 = isocitrate dehydrogenase 2, ATRX = α-thalassemia/mental retardation, X-linked gene; M = morphology; CC = cell count; p = proliferation; A = apoptosis. Please click here to download this Table.
Table 2: Medium compositions. H-GPSA = Hibernate A-Glutamax pencillin streptomycin amphotericin B. PDO = Patient-derived organoids DMEM: Dulbecco’s Modified Eagle’s Medium. NEAA: non-essential amino acids. Pen Strep: Penicillin/ Streptomycin Please click here to download this Table.
Table 3: Overview of the used techniques to generate cell culture models. Please click here to download this Table.
Supplementary Figure 1: Individual channels of proliferation staining of PDOs. (A) DAPI (blue), (B) GFAP-positive cells (green), (C) Ki67-positive cells (red) and (D) overlay channel in PDOs from patients 3, 4 and 5. Scale bars = 100 µm. Please click here to download this File.
Supplementary Figure 2: Individual channels of apoptosis staining of PDOs. (A) DAPI (blue), (B) GFAP-positive cells (green), (C) CC3-positive cells (red) and (D) overlay channel in PDOs from patients 3, 4 and 5. Scale bars = 100 µm. Please click here to download this File.
