Culturing Fluorescent Excitatory Neurons from a Transgenic Mouse Pup Brain
Abstract
Source: Turko, P., et al. Primary Cell Culture of Purified GABAergic or Glutamatergic Neurons Established through Fluorescence-activated Cell Sorting. J. Vis. Exp. (2019)
The video illustrates a technique for culturing fluorescent GABAergic and glutamatergic neurons from the cortex and hippocampal tissues of transgenic mouse pups. The process includes obtaining purified single-cell suspensions of neurons, sorting GABAergic neurons based on their fluorescence protein expression, and seeding them onto poly-L-lysine-coated plates to facilitate attachment and cell proliferation.
Protocol
All procedures involving sample collection have been performed in accordance with the institute's IRB guidelines.
1. Coating Glass Coverslips with Poly-L-lysine
- Prepare glass coverslips by defrosting a 5 mL aliquot of 200 µg/mL poly-L-lysine (PLL) solution. Dilute this stock solution to 20 µg/mL by adding 45 mL of pure injection grade water. Filter-sterilize the solution into a new 50 mL conical tube. Label this tube as "PLL Sterile".
NOTE: Use water stored at room temperature to speed up the defrosting process. - Add 100 sterile, round, 12 mm glass coverslips to the sterile PLL solution. Agitate the tube every 5–10 min, for 2–3 min, to ensure even coating. Coat the coverslips in this way for 1 h.
NOTE: German-manufactured round glass coverslips (diameter = 12 mm) are preferred, as they provide a reliable culture surface and fit easily into the wells of a 24-well cell culture plate. - After 40 min of PLL coating, take 2 pieces of tissue paper and lay them flat in the flow cabinet. Sterilize the paper using 70% ethanol, then flatten to remove creases and leave to dry.
- After coating the glass coverslips for 1 h with PLL, remove excess PLL solution and add sterile injection grade water. Gently agitate the coverslips for 2–3 s to allow removal of excess PLL. Repeat this rinsing step 2 additional times.
- Remove excess water, and then transfer the coverslips to the sterile tissue paper. Carefully separate each coverslip using curved forceps and leave to dry.
NOTE: Coverslips that do not easily separate can be discarded. Alternatively, a small amount of water can be added to help separation. - Once dry, transfer the coverslips to a 24-well culture plate.
NOTE: Coverslips should be made ready approximately 30 min–1 h before starting the dissection process. Plates can be stored in an incubator at 37 °C and 5% CO2 until required.
2. Dissociation of Hippocampal and Cortical Tissue
- Preparation of cell culture solutions
- For the dissociation of neural tissue, first defrost a 40 mL aliquot of cell culture buffer (composition [in mM]: 116 NaCl, 5.4 KCl, 26 NaHCO3, 1.3 NaH2PO4, 1 MgSO4·7H2O, 1 CaCl2·2H2O, 0.5 EDTA·2Na·2H2O and 25 D-glucose, pH = 7.4). Measure out 12 mL of cell culture buffer to a 15 mL conical tube; label as "BSA". Measure out 5 mL of cell culture buffer to a different 15 mL tube; label as "papain". Incubate both tubes in a water bath for 15 min at 37 °C.
- Filter-sterilize the remaining cell culture buffer, label as "Buffer – Sterile" and store at 4 °C until required.
- Weigh out 120 mg of Bovine Serum Albumin (BSA) and add to the tube labeled "BSA". Weigh out 7 mg of papain and add to the tube labeled "papain". Return both tubes to the water bath for 15 min.
NOTE: It is helpful to add a small amount of buffer to the weigh boat to facilitate transfer of the papain powder. - Once all powders have dissolved, filter-sterilize each solution to a fresh 15 mL conical tube. For tube papain, label the filtered solution as "papain – Sterile". For tube BSA, divide the sterile BSA solution into 3 tubes, each containing 4 mL of BSA solution. Label each tube as "BSA – Sterile" and either "1", "2" or "3". Return all tubes back to the water bath and continue to incubate at 37 °C until required.
- Preparation for tissue dissection
- Take a single piece of 35 mm filter paper (see Table of Materials) and sterilize with 70% ethanol; leave to dry in the lid of a 100 mm Petri dish, in a flow cabinet. Lay out the scissors, forceps, scalpel and spatula (see Table of Materials) required for the dissection of the hippocampus and cortex.
- Place two 35 mm Petri dishes and a 100 mm Petri dish containing sterile filter paper in an accessible location in center of the flow cabinet.
- Collect transgenic NexCre;Ai9 and VGAT Venus mouse pups that are to be dissected. Use a fluorescent lamp with appropriate excitation and emission filters (see Table of Materials) to discriminate fluorescent pups from wild type littermates.
- Immediately before dissecting animals, fill each 35 mm Petri dish with chilled, sterile cell culture buffer.
NOTE: One Petri dish is for cleaning the tools between dissections, and the other is for collecting the hippocampus and cortex.
- Tissue dissection
- As soon as the cell culture buffer is poured, decapitate a postnatal day 0–2 transgenic mouse or rat pup using large, sharp scissors and use a sterile 100 mm Petri dish to transfer the head into the flow cabinet. Use forceps, scissors and a spatula to carefully transfer the brain of a transgenic pup to sterile filter paper.
- Use a type 22 scalpel to dissect away the cerebellum and to separate the 2 hemispheres. Use a small spatula to roll the hemispheres laterally. On each hemisphere, press down gently so that the cortex comes into contact with the filter paper. Gently move midbrain regions to reveal the hippocampus and cortex.
NOTE: Be careful not to apply too much pressure when pressing down on the hemispheres or cells can become crushed. - Use a scalpel or spatula to separate the hippocampus and cortex from each hemisphere. Transfer dissected tissue to a 35 mm Petri dish containing chilled cell culture buffer.
NOTE: If dissecting multiple animals, store sterile cell culture buffer in a 50 mL conical tube on ice. After each dissection, transfer dissected tissue to the chilled cell culture buffer. - After successful dissection of cortico-hippocampal tissue, transfer the sterile papain solution from the water bath to the flow cabinet. Use a 3 mL Pasteur pipette to transfer the dissected hippocampus and cortex to the lid of a sterile 35 mm Petri dish. Chop in a criss-cross motion, using the flat part of a type 22 scalpel, until the tissue is in small pieces.
- Use a small amount of papain solution to transfer the chopped tissue from the lid of the Petri dish to the sterile papain tube. Return the papain tube to the water bath and incubate the tissue at 37 °C for 25 min.
- During papain incubation, make up complete fluorescence media solution. Defrost a 1 mL aliquot of B27, a 0.5 mL aliquot of Glutamax and a 0.5 mL aliquot of Pen-Strep. Aliquot 48.5 mL of Hibernate A low fluorescence media to a 50 mL conical tube. Add B27, Glutamax and Pen-Strep to the solution. Shake the solution well, then filter-sterilize and label as "Hibernate A complete media" (with date and initials). Incubate at 37 °C in a water bath.
- After papain incubation, transfer the papain-tube and sterile BSA-tubes to the flow cabinet. Use a 1 mL Pasteur pipette to remove only the cortico-hippocampal tissue from the papain tube into BSA-tube 1.
NOTE: Take care to minimize the transfer of excess papain solution.
- Dissociation of cells
- In order to break up any large clumps of tissue, triturate the tissue several times using a 1 mL Pasteur pipette. Following this, triturate the tissue 7 times using a fine tip Pasteur pipette. Leave the tissue to stand for 30 s, allowing larger pieces of tissue to sediment.
- After 30 s, transfer the lower 1 mL of solution and tissue from BSA-tube 1 to BSA-tube 2. Triturate the tissue in BSA-tube 2 several times using a fine tip Pasteur pipette.
- After trituration, transfer again the lower 1 mL of tissue and solution from BSA-tube 1, but now to BSA-tube 3. Triturate the tissue in BSA-tube 3 several times using a fine tip Pasteur pipette.
- After trituration, transfer all solution and tissue from tubes 2 and 3 into BSA-tube 1. Triturate a further 2-3 times and centrifuge at 3,000 x g for 3 min.
- During centrifugation, make up complete Neural Basal A media (NBA media). Aliquot 48.5 mL of NBA media to a 50 mL conical tube and incubate at 37 °C in a water bath. Label this tube "NBA only". In addition, defrost a 1 mL aliquot of B27, a 0.5 mL aliquot of Glutamax and a 0.5 mL aliquot of Pen-Strep at room temperature.
- Following centrifugation, carefully remove the supernatant from the pelleted tissue and re-suspend the cells in 2 mL of complete media. Use a P1000 pipette to triturate the tissue up and down 20 times.
NOTE: Take care not to pipette the cells too vigorously. If too much pressure is applied, the cells may become damaged. - Use a 1 mL Pasteur pipette to filter the cell suspension through a 30 µm cell sieve into a polystyrene sample tube.
NOTE: If the cell suspension does not pass immediately through the cell sieve, it may be necessary to press on top of the sieve with a sterile glove to start the flow. This important step prevents clogging of the cell sorter. - For collecting neurons after sorting, prepare collection tubes by pipetting 300 µL of complete media to the required number of polypropylene tubes.
- The cell suspension is now ready to be taken to the cell sorter for sorting. Take the cell suspension, extra collection tubes and spares complete media in case dilution of the sample is required.
3. Cell Sorting of Purified GABAergic or Glutamatergic Neurons
NOTE: To minimize the chance of bacterial contamination during sorting rinse the sample tubing of the sorter with 70% ethanol for at least 5 min prior to sorting. Detailed sorting parameters vary among instruments, fundamental considerations are as follows.
- During the first cell sort, establish levels of background fluorescence from unlabeled cells by sorting and comparing cells from a wild type animal.
- For each fluorescent cell type to be sorted, choose the appropriate excitation and emission filters. Excite Venus and TdTomato proteins using 488 nm or 531 nm excitation wavelengths, respectively. Detect emitted light through 530/40 and 575/30 emission filter sets, respectively.
- Add polypropylene collection tubes, containing 300 μL of complete media, to the cell sorter for cell collection.
- For high purity, sort brightly labeled fluorescent cells (see Figure 1B for example dot plots of sorted cells).
- Following sorting, for optimal results, perform a purity test of the sorted cells to estimate purity levels. Note that the typical recovery rate of cells following sorting is between 70–80%.
- Note down the number of cells sorted in each collection tube. This value is given by the cell sorting instrument.
NOTE: Cell sorting can be performed using a 70 µm nozzle (70 psi sheath fluid pressure) or a 100 µm nozzle (20 psi sheath fluid pressure).
4. Culturing of Sorted Neurons
- Following cell sorting, transfer collected cells to 2 mL round bottom centrifuge tubes, using a 1 mL Pasteur pipette. Centrifuge the cells at 3,000 x g for 3 min to form a cell pellet.
NOTE: To help identify the location of the cell pellet, orientate the round bottom centrifuge tubes with the hinge of the lid facing outward, which allows the cell pellet to form on the back of the centrifuge tube (i.e., on the same side of the tube as the hinge). - During centrifugation, add 1 mL of B27, 0.5 mL of Glutamax and 0.5 mL of Pen-Strep to the 48.5 mL of pre-warmed NBA media. Shake the solution well, then filter sterilize and label as "NBA complete media" (with date and initials). Return to water bath until required.
- Following centrifugation, use a 1 mL Pasteur pipette to transfer the supernatant to a different centrifuge tube (retain the supernatant incase the cell pellet was disturbed). Re-suspend the cell pellet in the required amount of pre-warmed complete NBA media to achieve a cell density of 1,000 cells/µL. Re-suspend the cells by pipetting up and down with a P200 or P1000 pipette.
- To confirm the presence of dissociated cells, check the cell solution under a microscope, using a 4x or 10x objective lens. Alternatively, pipette a small amount of cell solution onto a coverslip and check under the microscope. If large numbers of cells are present, the supernatant from step 4.3 can be discarded.
- Before plating the cells, vortex on a medium speed for 2–3 s to ensure an even cell suspension. Following vortexing, quickly pipette 10 µL of the cell suspension to the center of each coverslip. After pipetting the cells, quickly return each plate to the incubator (5% CO2/37 °C) and incubate for 1 h.
NOTE: If adding cells to multiple 24-well plates, ensure even cell densities by vortexing cells between plates. - After 1 h, feed the cells with 500 µL of pre-warmed complete NBA media and return to the incubator.
NOTE: When feeding the cells, it is important to pipette the media gently down the side of the well to avoid cell detachment. For short experiments (<16 days), feeding at the above density is not necessary. When plating a greater density of cells, more frequent feeding intervals may be required.
Representative Results
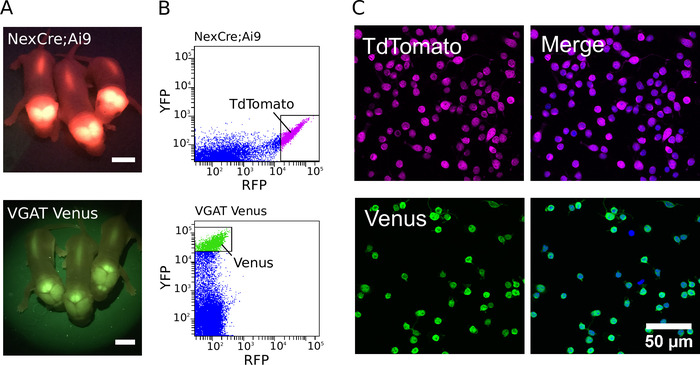
Figure 1: Purification of glutamatergic and GABAergic neurons. (A) Images showing fluorescent signal from the transgene expression of TdTomato in NexCre;Ai9 mice (top) and Venus in VGAT Venus mice (bottom). Scale bars = 5 mm. (B) Intensity scatter plots of the TdTomato and Venus fluorescence of cortico-hippocampal dissociated cells from NexCre;Ai9 mice (top) and VGAT Venus mice (bottom). Strongly fluorescent TdTomato or Venus neurons were selected for sorting (indicated by the gating boxes). (C, left) Confocal images of sorted TdTomato (top) and Venus (bottom) positive neurons. (C, right) Merged image showing cells co-stained with DAPI (in blue pseudocolour). TdTomato fluorescence is endogenous and remains strong despite fixation (in magenta pseudocolor). Venus expression is enhanced using a combination of a primary antibody directed against GFP and Alexa Fluor-488-conjugated secondary antibody (in green pseudocolor)
Divulgaciones
The authors have nothing to disclose.
Materials
| Neural Basal A media (NBA) | ThermoFisher Scientific | 10888022 | Cell Culture Buffer |
| B27 | ThermoFisher Scientific | 17504001 | Culture supplement |
| Glutamax | ThermoFisher Scientific | 35050-038 | Culture supplement |
| Penicillin Streptomycin (10,000 U/mL) | ThermoFisher Scientific | 15140-122 | Antibiotic |
| Poly-L-Lysine | SIGMA | P1399 | Coverslip coating |
| Papain | SIGMA | P4762-1G | Enzyme |
| Bovine Serum Albumin | SIGMA | A3294-100G | Serum |
| Hibernate A low fluorescence media | Brain Bits Ltd | HALF | Cell Transport media |
| Fine Tip Pasteur Pipette | Neo Labs | – | Used for trituration of cells |
| 24-well plates | BD | 353047 | Culture plate |
| 50 mL Falcon tubes | BD | 352070 | – |
| 15 mL Falcon tubes | BD | 352096 | – |
| Glass coverslips: 12 mm round | Roth | P231.1 | – |
| 35 mm Petri dish | Corning | 353001 | – |
| 100 mm Petri dish | Corning | 353003 | – |
| 30 µm CellTrics Cell Sieve | sysmex | 04-004-2326 | To remove cell clumps before cell sorting |
| Round bottom polystyrene tubes | BD | 352054 | Transport tube for sorted cells |
| Round bottom polypropylyne tubes | BD | 352063 | Collection tube for sorted cells |
| Extra fine Bonn Scissors | Fine Scientific Tools | 14084-08 | To remove overlying skin and bone of mice |
| Extra narrow Scissors | Fine Scientific Tools | 14088-10 | To remove overlying skin and bone of rats |
| Forceps | Fine Scientific Tools | 11242-40 | To hold the head in place |
| Spatula (130 mm long / 5 mm tip width) | Fine Scientific Tools | 3006.1 | To remove the brain to filter paper |
| Scalpel Blades | Swan-Morton | #0308 | To mechanically dissociate neural tissue |
| Haemocytometer (Neubauer Imroved) | Optik Labor | – | To cell count dissociated cells |
