Generation and Differentiation of Primary Neurospheres from Zebrafish Neural Stem Cells
Abstract
Source: Lopez-Ramirez, M. A. et al., Isolation and Culture of Adult Zebrafish Brain-derived Neurospheres. J. Vis. Exp. (2016)
This video demonstrates the generation and differentiation of primary neurospheres. Neural stem cells from zebrafish, in the presence of growth factors, proliferate into free-floating neurospheres with self-renewal capacity, which later differentiate into glial cells and neurons.
Protocol
1. Preparations
- Prepare 10 ml of dissection medium, and add 200 µl of 100x penicillin-streptomycin into 9.8 ml of Dulbecco's Modified Eagle Medium F12 (DMEM/F12).
- Prepare L-Cysteine solution: to 10 ml of water for tissue culture, and add 120 mg of L-cysteine. Store the L-cysteine solution at 4 °C for up to 2 weeks or in aliquots at -20 °C for up to 1 year.
- Prepare DNase I solution: to 1 ml of water for tissue culture, add 10 mg of DNase I. Store the DNase I solution at 4 °C for up to 2 months.
- Prepare papain solution: to prepare papain solution, add 100 µl papain (approximately 140 units), 100 µl DNase I (1%), and 200 µl L-cysteine (12 mg/ml) into 5 mL of DMEM/F12. Freshly prepare the papain solution every time and sterilize with a 0.22 µm pore size filter before use.
- Prepare washing solution: to prepare 100 ml of washing solution, add 650 µl glucose 45%, 500 µL 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, HEPES 1 M and 5 ml fetal bovine serum (FBS) into 93.85 ml Dulbecco's phosphate buffered saline (DPBS) 1x. Sterilize the washing solution with a 0.22 µm pore size filter before use. Store the washing solution at 4 °C for up to 2 months.
- Prepare insulin solution: to prepare 2 ml of insulin solution, add a 25 µl drop of 10 N sodium hydroxide (NaOH) and 100 mg insulin into 2 ml of water for tissue culture.
- Prepare epidermal growth factor (EGF) and fibroblast growth factor (FGF) solutions: Dissolve both mitogens in DMEM/F12 at 100 µg/ml concentration. Store as 10 µl aliquots at -20 °C.
- Prepare B-27 and N-2 media: store B-27 and N-2 supplements at -20 °C as 500 µl and 1 ml aliquots, respectively.
- Prepare Z-differentiation condition medium: to prepare 100 ml of Z-differentiation condition medium, add 40 µl insulin (50 mg/ml), 500 µl B-27, 1 ml N-2, 650 µl glucose 45% and 1 ml of 100 x penicillin-streptomycin into 97.81 ml of DMEM/F12. Sterilize the Z-differentiation condition medium with a 0.22 µm pore size filter before use. Store the Z-differentiation condition medium at 4 °C for up to 1 week.
- Prepare Z-condition medium: to prepare 50 ml of Z-condition media, add 10 µl of EGF and 10 µl of FGF into 50 ml of sterile Z-differentiation condition medium (20 ng/ml). Store the Z-condition medium at 4 °C for up to 1 week.
- Prepare coating solution for differentiation culture: for 5 ml extracellular coating solution, add 100 µl of the extracellular matrix solution (e.g., Matrigel) into 4.9 ml of DMEM/F12. Thaw extracellular matrix solution at 4 °C on ice. Once defrosted, keep at 4 °C for up to 2 weeks.
2. Dissection of the Adult Zebrafish Brain
- Prepare a dissection bed by filling a 100 mm x 15 mm Petri dish with gel ice packs. Then place the lid on the petri dish and incubate at -20 °C until the gel freezes. On top of the lid place a square of clean filter paper and wrap both the filter paper and petri dish with plastic film.
- Clean and sterilize all the microdissection instruments by 70% ethanol or heat before each use. Place all sterilized dissection instruments near the dissecting microscope and, right before euthanasia, place the dissection bed under the microscope with optical fiber illumination.
- Collect 2 adult zebrafish for a whole brain neurosphere preparation; and 3 to 4 zebrafish to generate neurospheres from dissected brain regions.
- Euthanize adult zebrafish (8-12 months old) using a protocol approved by the Institutional Animal Care and Use Committee. Next, immerse the fish in 75% ethanol for 5-10 sec and quickly place in the dissection bed followed by decapitation at the level of the gills using a surgical blade.
- To euthanize animals, administer an overdose (300 mg/L) of tricaine methane sulfonate until the animal's heartbeat gradually slows down and circulation stops, then immerse in iced water.
- Turn the head dorsal side down, and using the scissors make a longitudinal cut from the cut side to the mouth. Using the forceps expose the base of the skull and remove all the adjacent tissue. Cut the lateral walls of the skull from the beginning of the spinal cord towards the tectum.
- Using the scissors, cut and remove the optic nerves and then remove both sides of the most lateral part of the skull at the level of the tectum. Turn the head ventral side up. Using forceps, peel off the rest of the most apical part of the skull.
- Transfer the brain along with the remaining part of the skull into a new dish with the dissection medium (1.1). Clean the brain tissue in the dissection medium using the micro knife's plastic handle, keeping all brain structures intact.
- From this point, continue the protocol using the whole zebrafish brain.
- Alternatively adapt this protocol to specific brain regions to generate neurospheres from the whole zebrafish brain or from the telencephalon, tectum/diencephalon or cerebellum dissected with a fresh scalpel. Use a neural specific fluorescent zebrafish neural transgenic line to dissect the brain region of interest according to Figure 1.
3. Single Cell Dissociation of Adult Brain
- Perform all subsequent work in a biological safety cabinet. Transfer the brain tissue into a 1.5 ml tube containing 800 µl of dissection medium (prepared in step 1.1). Remove the dissection medium by pipetting, without touching the brain tissue.
- Add 500 µl of papain solution (step 1.4) and digest the brain tissue at 37 °C for 10 min. Do not incubate longer than 15 min as it could decrease cell viability.
- After incubation, transfer the brain tissue with 500 µl of the papain solution into a 15 ml conical tube using a 1,000 µl pipet tip cut at ~0.25 inches from the bottom. Gently dissociate the brain tissue by slowly pipetting up and down 10 times with the same tip. Do not pipet up and down more than 15 times and do not generate air bubbles during this step, as it could alter cell viability.
- Incubate the brain tissue again at 37 °C for 10 min followed by pipetting up and down 10-13 times using an uncut 1,000 µl regular tip. Do not pipet up and down more than 15 times, to avoid cell death.
- Stop the enzymatic digestion by adding 2 ml of washing solution (step 1.5) and centrifuge the cell suspension at 800 x g for 5 min at room temperature (RT). Carefully decant the supernatant and resuspend the pellet in the remaining solution by tapping the tube vigorously with a finger and then by pipetting up and down with a 1,000 µl regular tip. Next, add 2 ml of washing solution.
- At this stage, check under the microscope that a single cell suspension has been obtained. Centrifuge the cell suspension again at 800 x g for 5 min at RT. Remove the supernatant and re-suspend the pellet using 1 ml of fresh Z-condition medium (step 1.10).
- Stain cells by using trypan blue and count the living cells using a hemocytometer by excluding blue dead cells. Prepare a 24-well plate with 200 µl of cell suspension and 300 µl of fresh Z-condition medium in each well. Seed cells at a density of ~500 cells/µl. Maintain cultured cells in an incubator at 30 °C in 5% CO2, since zebrafish brain-derived neurospheres do not grow well at 37 °C.
4. Generation of Primary Neurospheres
- After 1 day in vitro (DiV1), observe the single cell suspension obtained from the adult whole brain under the microscope and note whether debris has accumulated in the center of the well. Carefully remove any debris by pipetting approximately 100 µl of medium (less if possible). Next add 100 µl of fresh Z-condition medium. Single cell suspensions should be observed at this time (Figure 2A DiV1).
- After 2 days in vitro (DiV2), expand the cultured cells. Using a 1,000 µl pipette with the tip cut, collect and transfer 250 µl of cell suspension from each well into a new well. Add 250 µl of fresh Z-condition medium into all wells. Before expanding the cultured cells, homogenize the suspension very gently by pipetting up and down throughout the well.
- Repeat steps 4.1 and 4.2 for the following 4 days in vitro (DiV4). A progressive increase in size of neurospheres should be visible at the center and edges of the well at DiV3 and DiV4 (Figure 2B and C).
NOTE: Primary neurospheres can be processed for passage or differentiation to assess multipotency. Alternatively, they can be processed for nucleofection or lipo-transfection to characterize the role of specific genes during the differentiation process.
5. Passaging of Primary Neurospheres
- Remove 250 μl of tissue culture from each well and mechanically dissociate DiV4 neurospheres with a 1 ml pipette. Do not pipet up and down too rapidly as air bubble formation may increase cell death.
- Count the cells with a hemocytometer and distribute 800 cells/µl in 250 µl of primary culture supernatant into each well of a 24-well plate.
- Add 250 µl of Z-condition medium to each well.
- After 2 days in vitro (DiV2), expand the cultured cells as reported in step 4.2 and 4.3.
6. Differentiation of Primary Neurospheres
- Sterilize cover glasses by immersion in 70% ethanol for 10 min, then dry cover glasses at RT by placing them into each well of a 12-well plate.
- Add 300 µl of extracellular coating solution (step 1.11) at the center of the cover glass in such a way that it can expand widely and cover the entire cover glass. Then, incubate at 37 °C for 1 h (using the humidified cell culture incubator).
- Remove the extracellular coating solution after incubation, and dry the cover glass for 2 hr at 37 °C.
- Remove 250 µl of tissue culture from each well. Mechanically dissociate all the DiV4 primary neurospheres per well with a 1 ml pipette (with wider-end tip) by pipetting up and down.
- Seed dissociated neurosphere cell suspensions at a cell density of 4 x 103 cells/ml on previously prepared extracellular matrix-coated cover glasses (step 6.1-6.3) and keep for at least 30 min, at 30 °C in 5% CO2, until attached to the substrate. Then, remove the culture medium and rapidly add 1 ml of pre-warmed fresh Z-differentiation condition medium (step 1.9) before culturing the cells for 24 hr at 30 °C in 5% CO2.
- Every two days, replace half of the Z-differentiation condition medium during the time of cell differentiation.
Representative Results
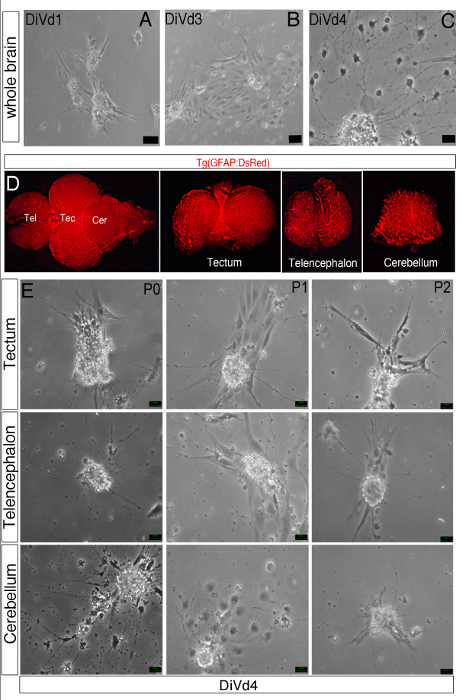
Figure 1: Differentiation of Zebrafish Brain-derived Neurospheres. (A–C, E) Phase contrast images of whole brain-derived neurospheres cultured in the Z-differentiation medium during 1 (A, DiVd1), 3 (B, DiVd3) and 4 (C and D, DiVd4) days. (E) Dorsal view of the whole brain of a 12 month old Tg(GFAP:DsRed) zebrafish. The telencephalon (Tel), tectum (Tec) and cerebellum (Cer) were dissected and collected as shown. (D) Images of DiVd4 neurospheres derived from the tectum, telencephalon and cerebellum at Passage 0 (P0), 1 (P1) and 2 (P2). Scale bar: 25 µm.
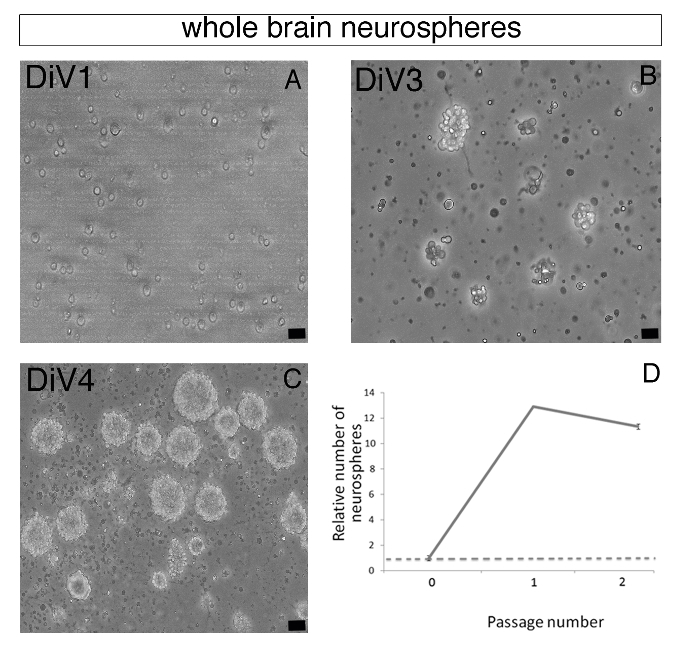
Figure 2: Forming Zebrafish Brain-derived Neurospheres. (A–C) Representative phase contrast images of whole brain-derived primary floating neurospheres were observed on day 1 (DiV1, A), day 3 (DiV3, B), and day 4 (DiV4, C). (D) Chart representing the relative number of neurospheres at Passages 1 and 2 compared to the neurosphere number at Passage 0. After Passage 2, neurosphere formation was drastically decreased. Scale bar: 25 µm.
Divulgaciones
The authors have nothing to disclose.
Materials
| DPBS 1X | Life Technologies | 14190-144 | |
| DMEM/F12 1X | Life Technologies | 11330-032 | |
| L-Cysteine hydrochloride monohydrate | Sigma | C6852-25g | |
| B-27 | Life Technologies | 17504-044 | |
| N-2 | Life Technologies | 17502-048 | N-2 supplement (100x) liquid |
| HEPES | Life Technologies | 15630 | 1M |
| D-(+)-Glucose 45% | Sigma | G8769 | |
| Penicillin-streptomycin | Life Technologies | 15140-122 | |
| Fetal Bovine Serum | Life Technologies | 16000044 | |
| Human FGF-basic | Peprotech | 100-18B | |
| Human EGF | Peprotech | AF-100-15 | |
| Insulin | Sigma | I5500-50 mg | |
| DNAse | Sigma | DN25-10mg | |
| Papain | Worthington Biochemical Corporation | LS003126 | |
| Matrigel | Becton Dickinson | 356234 | |
| PFA | TCI | P0018 | |
| PBS | AmericanBio | AB11072-04000 | |
| Tricaine MS-222 | Sigma | A5040 | Stock solution of 4 mg/ml. |
| Trycold gel | Sigma | TGP8 | gel pack |
| Trypan blue stain | Life Technologies | 15250061 |
