We have presented a five day protocol for the preparation of an endogenous Escherichia coli based TX-TL cell-free expression system. A sample timeline for creating the reagents – crude cell extract and buffer – can be found in Figure 1. Once created, these can be stored at -80 °C for up to one year. After reagents are created, experimental setup and execution can be done in less than 8 hr.
In addition, we optimized the expression conditions of the TX-TL cell-free expression system. Other user-supplied additions, such as buffers or DNA solutions, should be calibrated for toxicity beforehand. For example, different methods of processing plasmids result in different expression due to salt content. We also tested the effect of Tris-Cl elution buffer on reaction efficiency (Figure 5).
An example of crude cell extract calibration, referring step 4.1 to 4.9, is shown in Figure 4a. In general, our experiments show that the crude cell extract is most sensitive to Mg-glutamate levels, followed by K-glutamate levels. To demonstrate the cell-free expression system, we constructed and tested a negative feedback loop based on tet repression.26 (Figure 6). In the cell-free expression system, the same circuit run with and without aTc shows a 7-fold end-point expression change of deGFP reporter after eight hours of expression. Although this experiment does not require global inducers or repressors, if necessary they can be added under “Master Mix Preparation.”
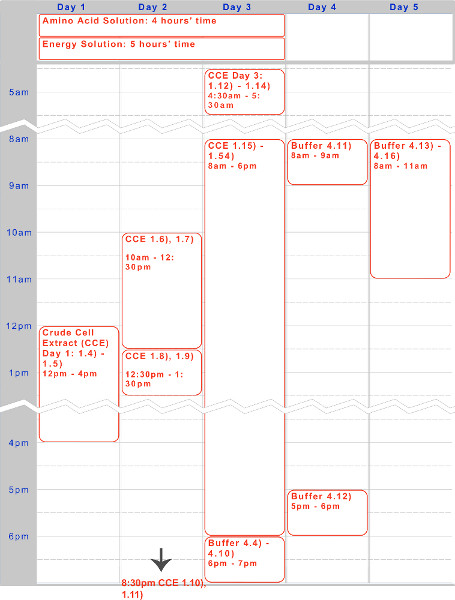
Figure 1. Timeline for crude cell extract, amino acid solution, and energy solution preparation. A five-day timeline for a typical execution of the protocol is given above, optimized for overnight incubations and daytime working steps.
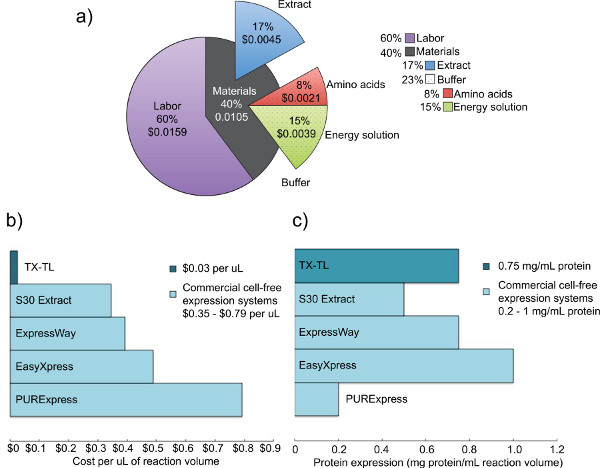
Figure 2. Cost and expression analysis of competing crude cell extracts. a) Breakdown of the costs of labor and materials of the TX-TL cell-free expression system. Based on costs of reagents as of December 2012, and labor costs of $14 per hour. b) Comparison of TX-TL cell-free expression system costs vs. other commercial systems. Costs are broken down per μl, although reaction volumes may vary per kit. c) Comparison of TX-TL cell-free expression system yield vs. other commercial systems. Protein expression yield determined by manufacturer standards. Click here to view larger figure.

Figure 3. Loading and processing of a bead-beating tube. a) Demonstration of correct viscosity of cell-bead solution. Cell-bead solution will have a viscosity dependent on many factors, including amount of S30A buffer added, amount of beads added, and time spent on ice. b) Loading of bead-beating tube before quick tabletop centrifugation. The centrifugation removes bubbles accumulated during loading. c) Bubbles surfacing after tabletop centrifugation. The size of the bubbles will vary; they can be popped or removed using a pipette tip. d) Completely filled bead-beating tube before capping. A meniscus is formed in the bead-beating tube, and the cap has enough to cover and cause small amounts to overfill. e) Correctly loaded filter apparatus. These can be reused. f) Comparison of correctly vs. incorrectly processed bead-beating tube. The tube on the left is a well-beat tube – it features a small and well-delineated top layer, and very clear supernatant. The tube on the right is suboptimal, based on the larger, hazy second layer and the hazy supernatant. Tubes that are suboptimal should not undergo additional processing.
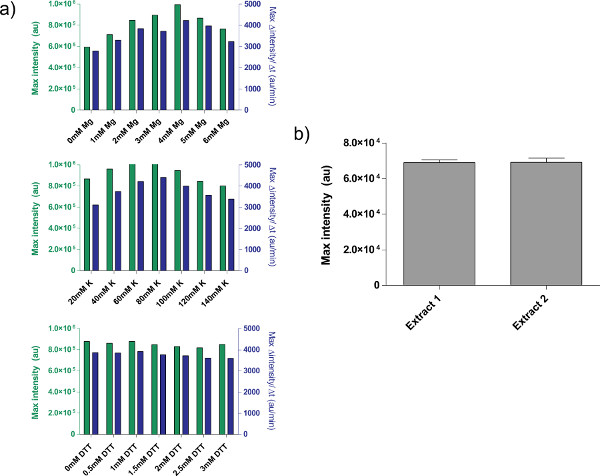
Figure 4. Properties of crude extract preparations. a) Typical calibration plots for crude cell extract. Crude extract is calibrated for additional Mg-glutamate, K-glutamate, and DTT levels, in that order. Shown is endpoint fluorescence after 8 hr, as well as maximal rate of protein production based on a 12-minute moving average. Based on these plots, an acceptable range of additional Mg-glutamate is 4 mM, K-glutamate is 60-80 mM, and DTT is 0-3 mM. Note that every crude extract needs to be calibrated independently for these three variables. b) Variation from extract preparations. Endpoint fluorescence of two crude extracts prepared on different dates is shown; error bars are 1 standard deviation from three independent runs on different days. Click here to view larger figure.
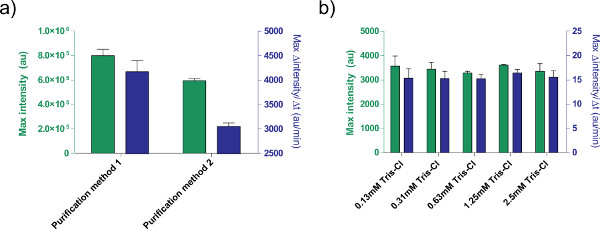
Figure 5. Effects of DNA solution on expression efficiency. a) Comparison of two different purification methods for processing plasmids. 1 nM of pBEST-OR2-OR1-Pr-UTR1-deGFP-T500 is prepared using only a QiaPrep Spin Miniprep Kit (Purification method 1) or post-processed with a QiaQuick PCR purification kit (Purification method 2). Shown is endpoint fluorescence after 8 hr, as well as maximal rate of protein production based on a 12-minute moving average. Error bars are 1 standard deviation from four independent runs on different days. b) Effect of elution buffer (Tris-Cl). Different concentrations of Tris-Cl are compared in a cell-free expression reaction based on the expression of 1 nM of pBEST-OR2-OR1-Pr-UTR1-deGFP-T500. Concentrations given are final concentrations of Tris-Cl in the reaction; elution buffer used is 10 mM Tris-Cl. Error bars are 1 standard deviation from three independent runs on different days. Click here to view larger figure.
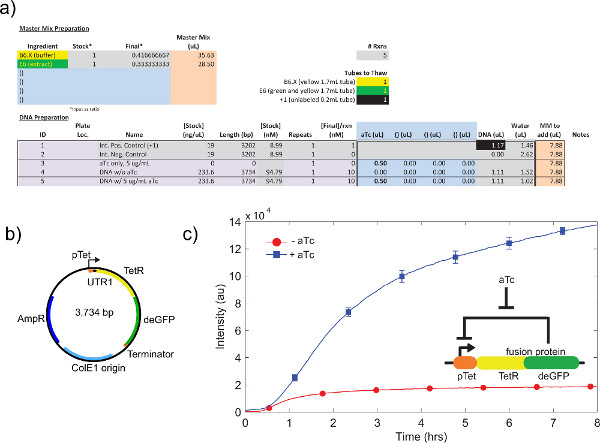
Figure 6. Sample TX-TL run of a negative feedback loop. a) Sample setup of a cell-free execution reaction. Tests “on” vs. “off” state of the negative feedback loop, with positive and negative controls. b) Plasmid map of negative feedback loop. c) Representative results. Data reflects experiment in a) and b), with negative control subtracted from signal. Genetic circuit shown in insert. Error bars are 1 standard deviation from three independent runs on different days. Click here to view larger figure.
| Name | Concentration | Amount | Sterilization | Notes |
| Chloramphenicol (Cm) | 34 mg/ml in ethanol | 1 ml | Filter sterilize (0.22 μM) | Can be made in larger volumes stored at -20 °C for later use. |
| 2xYT+P+Cm agar plate | 31 g/L 2xYT, 40 mM potassium phosphate dibasic, 22 mM potassium phosphate monobasic, 34 μg/ml chloramphenicol | 1 plate | Autoclave | |
| 2xYT+P media | 31 g/L 2xYT, 40 mM potassium phosphate dibasic, 22 mM potassium phosphate monobasic | 4 L | Autoclave |
Table 1. Reagents for day 1 of Crude Cell Extract protocol.
| Name | Concentration | Amount | Sterilization | Notes |
| Tris base | 2 M | 250 ml | Filter sterilize (0.22 μM) or autoclave | Can be stored at room temperature. |
| DTT | 1 M | 6 ml | Filter sterilize (0.22 μM) | Can be made in larger volumes and stored at -20 °C for later use. |
| S30A buffer | 14 mM Mg-glutamate, 60 mM K-glutamate, 50 mM Tris, pH 7.7 | 2 L | Autoclave | To reach pH 7.7, titrate with acetic acid. Add DTT to 2 mM final concentration just before use. Store at 4 °C. |
| S30B buffer | 14 mM Mg-glutamate, 60 mM K-glutamate, ~5 mM Tris, pH 8.2 | 2 L | Autoclave | To reach pH 8.2, titrate with 2M Tris. Add DTT to 1 mM final concentration just before use. Store at 4 °C. |
Table 2. Reagents for day 2 of Crude Cell Extract protocol.
| Falcon | ||||
| 1 | 2 | 3 | 4 | |
| Empty 50 ml Falcon (g) | ||||
| 50 ml Falcon with pellet (g) | ||||
| Pellet mass (50 ml Falcon with pellet – empty 50 ml Falcon) (g) | ||||
| S30A buffer volume to add (pellet mass* 0.9) (ml) | ||||
| Total mass of beads to add (pellet mass * 5.0) (g) | ||||
Table 3. S30A buffer and bead mass calculator, for day 3 of Crude Cell Extract protocol.
| Name | Concentration | Amount | Sterilization | Notes |
| HEPES | 2 M, pH 8 | 4 ml | None | To reach pH 8, titrate with KOH. |
| Nucleotide Mix | 156 mM ATP and GTP, 94 mM CTP and UTP, pH 7.5 | 1.5 ml | None | To reach pH 7.5, titrate with KOH. |
| tRNA | 50 mg/ml | 600 μl | None | |
| CoA | 65 mM | 600 μl | None | |
| NAD | 175 mM, pH 7.5-8 | 300 μl | None | To reach pH 7.5-8, titrate with Tris at 2 M. |
| cAMP | 650 mM, pH 8 | 200 μl | None | To reach pH 8, titrate with Tris at 2 M. |
| Folinic Acid | 33.9 mM | 300 μl | None | Although only 300 μl is needed, recipe in supplemental is for 1.15 ml. |
| Spermidine | 1 M | 150 μl | None | Store at 4 °C, heat to 37 °C to melt. |
| 3-PGA | 1.4 M, pH 7.5 | 3.2 ml | None | To reach pH 7.5, titrate with Tris at 2 M. |
Table 4. Reagents to prepare for Energy Solution protocol.
Supplemental Material 1. Recipes for Items.
Chloramphenicol, 34 mg/ml: Prepare 0.51 g chloramphenicol and add ethanol to 15 ml. Filter sterilize (0.22 μM), aliquot to 1 ml tubes, store at -20 °C for later use.
2xYT+P+Cm agar plate: Prepare 1.24 g 2xYT, 1.6 ml potassium phosphate dibasic solution @ 1 M, 0.88 ml potassium phosphate monobasic solution @ 1 M, 0.6 g agar, and water to 40 ml. Autoclave. Let cool to 50 °C and add 40 μl Cm. Aliquot 25 ml into a 100 x 15 mm Petri dish, and let cool for an hour.
2xYT+P media: Prepare 124 g 2xYT, 160 ml potassium phosphate dibasic solution @1 M, 88 ml potassium phosphate monobasic solution @ 1 M, and water to 4 L. Aliquot out into 2 x 1.88 L and 0.24 L. Autoclave.
Tris base, 2 M: Prepare 60.57 g Tris base and water to 250 ml. Sterilize, store at RT for later use.
DTT, 1 M: Prepare 2.31 g DTT and water to 15 ml. Filter sterilize (0.22 μM), aliquot to 1 ml tubes, store at -20 °C for later use.
S30A buffer: Prepare 10.88 g Mg-glutamate and 24.39 g K-glutamate, 50 ml Tris at 2M, acetic acid (to pH 7.7), and water to 2 L. Autoclave, store at 4 °C, add 4 ml 1 M DTT before use.
S30B buffer: Prepare 10.88 g Mg-glutamate and 24.39 g K-glutamate, Tris at 2 M (to pH 8.2), and water to 2 L. Autoclave, store at 4 °C, add 2 ml 1 M DTT before use.
HEPES: Prepare 1.91 g HEPES (MW 238.21), KOH (to pH 8), and water to 4 ml.
tRNA: Prepare 30 mg of tRNA and water to 600 μl.
CoA: Prepare 30 mg of CoA (MW 767.53) and water to 600 μl.
NAD: Add 34.83 mg of NAD (MW 663.43), Tris at 2 M (to pH 7.5-8), and water to 300 μl. (Add 27 μl of Tris at 2 M to bring the solution to pH 7.5-8).
cAMP: Add 42.80 mg of cAMP (MW 329.22), Tris at 2 M (to pH 8), and water to 200 μl. (Add 73 μl of Tris at 2 M to bring the solution to pH 8).
Folinic Acid (33.9 mM): To 20 mg of solid folinic acid calcium salt (MW 511.5), add 1.15 ml water.
Spermidine: Prepare 23.55 μl of spermidine (MW 145.25) and water to 150 μl. Prepare at room temperature after melting briefly at 37 °C.
3-PGA: Add 1.03 g of 3-PGA (MW 230.02), Tris at 2 M (to pH 7.5), and water to 3.2 ml. (Add 1.73 ml of Tris at 2 M to bring the solution to pH 7.5).
Nucleotide Mix: Add 145 mg of ATP dipotassium salt dihydrate (MW 619.4), 133 mg of GTP disodium salt (MW 567.14), 79.4 mg of CTP disodium salt dihydrate (MW 563.16), 82.6 mg of UTP trisodium salt dihydrate (MW 586.12), KOH at 15% dilution (to pH 7.5), and water to 1.5 ml. (Add 353 μl of KOH at 15% dilution to bring the solution to pH 7.5).
Supplemental Material 2. Bradford Assay.
- Remove Bradford agent from 4 °C and set at room temperature.
- Prepare 50 μl BSA Standard at 1 mg/ml and at 0.1 mg/ml.
- Prepare 40 μl 20x dilution of extract from step 1.47.
- Add 800 μl water to 7 cuvettes.
- Prepare standard cuvettes for 0 mg/ml, 1 mg/ml (10 μl 0.1 mg/ml BSA), 2 mg/ml (20 μl 0.1 mg/ml BSA), 4 mg/ml (4 μl 1 mg/ml BSA), 6 mg/ml (6 μl 1 mg/ml BSA).
- Prepare experimental cuvettes for 2 μl of sample and 4 μl of sample.
- Add 200 μl of Bradford agent to each cuvette and mix well by pipetting. Incubate at room temperature for at least 10 min.
- Produce standard curve at OD 595 nm using cuvettes from step 6.5. Reject standard curve if r2 < 0.95.
- Determine extract concentration at OD 595 nm using cuvettes from step 6.6.
Supplemental Material 3. Buffer calibration spreadsheet.
See TXTL_e(template)_calibration_JoVE.xlsx.
Supplemental Material 4. Cell-free expression run spreadsheet.
