NOTE: An assay timeline is provided in Figure 1.
1. Preparation of RNAi nematode growth medium (RNAi-NGM) plates
- Streak out E. coli HT115(DE3) containing either GFP or control RNAi vectors from glycerol stocks onto Luria-Bertani (LB) agar plates supplemented with 100 µg/mL ampicillin. Incubate the bacteria overnight at 37 °C.
- The following day, prepare RNAi-NGM plates (1.7% [w/v] agar, 0.3% [w/v] NaCl, 0.25% [w/v] peptone, 1 mM CaCl2, 5 µg/mL cholesterol, 25 mM potassium phosphate buffer [pH 6.0], 1 mM MgSO4, 25 µg/mL carbenicillin, and 5 mM isopropyl β-d-1-thiogalactopyranoside [IPTG]) according to the standard protocol18.
- For each strain in the assay, prepare a minimum of six RNAi-NGM plates seeded with GFP RNAi (two plates for each of the three biological replicates) and two RNAi-NGM plates seeded with control RNAi (one biological replicate).
- Loosely tent the plates with foil to protect from light and leave overnight at room temperature to dry.
- On the same day as RNAi-NGM plate pouring, prepare 4 mL of liquid cultures of RNAi bacteria.
- Under sterile conditions, aliquot 4 mL of LB broth supplemented with 10 µg/mL tetracycline and 100 µg/mL ampicillin into culture tubes. Pick single colonies from the LB agar plates into each tube and incubate overnight while shaking for approximately 16 h at 37 °C.
- Seed RNAi bacteria onto the RNAi-NGM plates.
- After overnight growth, measure the optical density (OD600) of cultures using a 1:5 dilution of bacteria with LB broth.
- Dilute RNAi bacteria to a relative OD600 of 2 using LB broth supplemented with 10 µg/mL tetracycline and 100 µg/mL ampicillin.
- Under sterile conditions, add 150 µL of the control or GFP RNAi bacteria onto each RNAi-NGM plate.
- Tent the plates with foil and allow the bacteria to grow for at least 24 h at room temperature before use.
NOTE: Unused RNAi-NGM plates can be stored inverted for up to 1 week at 4 °C in a sealed container in the dark.
2. Starting the RNAi inheritance assay: bleaching and plating embryos for the P0 generation
NOTE: Before starting the RNAi Inheritance assay, worms containing the GFP reporter mjIs134 [mex-5p::gfp-h2b::tbb-2 3'UTR]7 should be maintained unstarved at 21 °C for at least two generations.
- At 4 days (96 h) prior to the start of the RNAi inheritance assay (Figure 1), pick three or four gravid adults onto each 35 mm standard NGM plate seeded with OP50-1 bacteria.
NOTE: Three NGM plates per strain is sufficient. For strains with compromised fertility, more than four animals per plate may be required. - After 4 days, ensure that the adult population have started laying embryos on the plate. Wash the worms off the plates into 1.5 mL tubes using 800 µL of M9 buffer supplemented with Triton X-100 (TX-100) (22 mM KH2PO4, 42 mM Na2HPO4, 86 mM NaCl, 1mM MgSO4, 0.01% (v/v) TX-100). Repeat the wash and pool into the same tube.
- Isolate the embryos from the population by bleaching as follows.
- Prepare a bleaching solution with bleach (6% sodium hypochlorite) and 10 N NaOH in a 1.5:1 volume ratio. Prepare a sufficient quantity such that 250 µL can be used for each sample.
- Centrifuge the 1.5 mL tubes with the worms at 1,000 x g for 1.5-2 min.
- Aspirate the supernatant, leaving 100 µL without disturbing the worm 'pellet'. Alternatively, the tubes can be left for approximately 2 min to allow the worms to settle before aspirating.
- Add 650 µL of ddH2O to each tube to achieve a final volume of 750 µL.
NOTE: The following steps are time sensitive. - Add 250 µL of the bleaching solution to each tube and start a timer.
- Every 1-2 min, vortex the tubes thoroughly. After 5 min, check the worms under the stereoscope to monitor the degradation of adults.
- Continue vortexing until the worms are completely dissolved and only embryos remain. Immediately centrifuge the tubes at 1,000 x g for 1.5 min.
NOTE: Bleaching is generally complete within 6-7 min, but should be monitored under a stereoscope at a magnification sufficient to observe the released embryos (40x). Do not leave the worms in bleach for an extended period, as this will compromise the embryos. - After centrifugation, aspirate the supernatant and leave approximately 50-100 µL. Add 1 mL of M9 buffer with TX-100 to wash and mix by vortexing. Centrifuge again at 1,000 x g for 1.5-2 min and wash at least two more times.
- Aspirate the supernatant from the final wash and leave approximately 100 µL in the tube. Mix by vortexing. Pipette 2 µL from each tube twice onto a labelled glass slide. Count the embryos using a tally counter to estimate the concentration of embryos per microliter.
NOTE: Slides with multiple frosted wells can be used here to count replicates of multiple strains. Counting slides can be washed and reused.
- To start a semi-synchronized population growth, mix and pipette a volume containing 250 embryos onto each RNAi-NGM plate. Use two plates for each replicate. Allow the liquid to absorb.
- For the P0 generation treated with RNAi, incubate at 21 °C for 4 days (96 h) (from embryos into adulthood). Timings may vary with genotype.
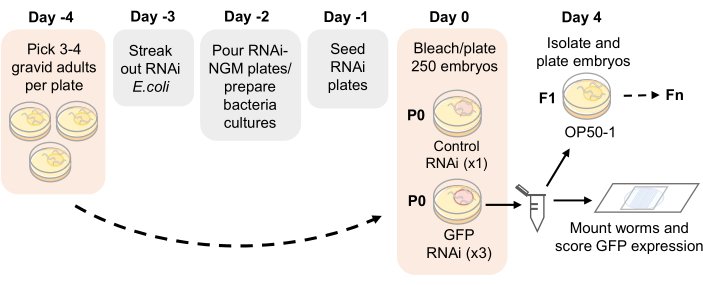
Figure 1: RNAi inheritance assay schematic. Proposed timeline for RNAi-NGM plate preparation and RNAi inheritance assay setup. Gravid adults are picked on Day -4 onto NGM plates seeded with OP50-1. After 4 days, adult progeny are bleached and embryos are plated onto RNAi-NGM plates. The P0 generation is exposed to RNAi for 4 days at 21 °C. Once the worms reach adulthood, replicates are passaged by bleaching and scored for germline GFP expression each generation. Please click here to view a larger version of this figure.
3. Passaging and scoring each generation for germline GFP expression
NOTE: To facilitate scoring, use a vinyl record to create agarose pads with linear ridges to line up the worms. This method was adapted from Rivera Gomez and Schvarzstein19.
- Prepare 1% (w/v) agarose by dissolving agarose in ddH2O in a small flask by heating in a microwave. Add a stir bar and cover with foil. If remelting, heat the agarose on a hotplate at 200 °C while stirring. Once melted, reduce the heat to 80 °C.
- Wash the worms off the NGM plates using 800 µL of M9 buffer with TX-100 into a 1.5 mL tube. Wash the plates twice to remove all the gravid adults. Check the plates under the stereoscope to confirm the animals have been collected efficiently.
- Centrifuge the tubes at 1,000 x g for 1.5-2 min or allow worms to settle for 2 min. Aspirate the supernatant and leave 100 µL without disturbing the worm 'pellet'.
- Prepare agarose pads for mounting the worms as follows.
- Place a drop of melted 1% (w/v) agarose onto the vinyl record (described previously19) using a glass Pasteur pipette (with the narrow end broken off).
- Orient a glass slide so that lines on the record are either horizontal or vertical and place the slide quickly onto the drop of agarose. Wait approximately 30 s before removing the glass slide from the record.
NOTE: If scoring multiple genotypes, it may be useful to make a larger agarose pad on one slide and cut it in half using a blade.
- Mount the worms onto the agarose pads.
- Under the stereoscope, add approximately 5 µL of 5 mM levamisole to the agarose pad. The levamisole dilution should be made fresh each week.
- Gently flick the tubes of worms to mix and transfer 5-10 µL of worms onto the agarose pad. Estimate the number of worms by eye as they are being added so that there are approximately 40 worms per replicate.
- Line up the worms in rows using an eyelash pick for easier scoring. Add a glass coverslip.
NOTE: Slides with animals mounted in this way can last for several hours before drying out.
- Isolate the embryos from the remaining animals using the bleaching protocol (refer to step 2.3) before scoring the slides. Plate 250 embryos onto 35 mm NGM plates seeded with OP50-120. Once the liquid is absorbed, invert the plates and return them to the 21 °C incubator.
- Use a florescence stereoscope with a GFP filterset. Count and record the number of GFP positive and GFP negative worms on the slides for each replicate using a tally counter.
NOTE: GFP positive worms will have nuclear GFP expression in their germline and embryos in utero. The germlines of GFP negative worms will not fluoresce, however, as the GFP starts to turn back ON in later generations, the fluorescence may be dim. It may be helpful to mount additional non-transgenic worm strains to account for any autofluorescence observed. - Passage the population by bleaching, as described above, approximately every 4 days (96 h) at 21 °C. Alternatively, passaging can be done on an alternating 3-day/4-day schedule for convenience. Plate an extra ~50 embryos for the shorter passaging generations if alternating, as there may be a lower yield of embryos after 72 h.
NOTE: Keep the P0 generation passaging time consistent at 4 days post-plating embryos.
4. Animal collection for ChIP
NOTE: The number of animals and timing depends on the strain, developmental stage, epitope, and number of immunoprecipitation (IP) targets. In the example below, collect animals for three IPs: H3K9me3, histone H3, and IgG control. The ChIP protocol was adapted from Askjaer et al.21.
- Prior to ChIP sample collection, expand the previous generation of the RNAi inheritance assay by an additional three NGM plates (to at least four plates per replicate). Grow for 4 days at 21 °C.
- Bleach the gravid adults to isolate the embryos (refer to step 2.3).
- For each strain, plate approximately 3,500 embryos onto 14 plates (250 per plate) and grow for 3 days at 21 °C to the young adult stage.
- Wash the animals into 1.5 mL tubes with phosphate-buffered saline (PBS) containing 0.01% (v/v) TX-100 (PBS/TX). Centrifuge at 1,000 x g for 2 min. Aspirate and pool the animals from one strain into one tube and wash three more times with at least 1 mL of PBS/TX.
- Aspirate to 1,000 µL, invert to mix, and count the number of animals in 3 µL three times (refer to step 2.3.9).
- Calculate the concentration of animals and transfer a volume corresponding to 3,000 animals to a new tube for formaldehyde crosslinking.
- Ensure that the total volume is at least 10 times the worm pellet volume.
5. Formaldehyde crosslinking
CAUTION: Work with formaldehyde in a fume hood to prevent vapour exposure.
- Add formaldehyde (1.8% final concentration) to the tube containing the worms. Rotate at room temperature for 6 min.
- Immediately freeze in liquid nitrogen. The experiment can be paused at this step and the samples can be stored at -80 °C.
- Thaw the crosslinked sample in a room temperature waterbath for 3 min and rotate for 16 min at room temperature.
- Add 1.25 M glycine to a final concentration of 125 mM and rotate for 5 min.
NOTE: Until magnetic bead elution, keep the samples on ice or at 4 °C and use ice-cold buffers. - Centrifuge the sample at 1,000 x g for 3 min. Wash three times with 1 mL of PBS/TX each time. Wash twice with 1 mL of resuspension buffer (150 mM NaCl, 50 mM HEPES-KOH [pH 7.5], 1 mM ethylenediaminetetraacetic acid [EDTA], 0.01% TX-100, protease inhibitor [one tablet per 5 mL]).
- After the last wash, leave enough buffer to ensure the total volume is at least three times the pellet volume and a minimum of ~100 µL.
6. Sonication
NOTE: Sonication parameters depend on the type and model of sonicator and animal stage. Parameters such as sample volume and concentration, on/off intervals, number of cycles, and power setting need to be optimized empirically. For example, over a time course of sonication, monitor worm lysis using a protein assay and determine when the concentration reaches a plateau. In addition, monitor when the average shear size of the genomic DNA is approximately 200-1,000 bp by electrophoresis of DNA, purified following crosslinking reversal, on a 1.5% agarose/tris-acetate-EDTA (TAE) gel.
- Measure the volume of samples from the previous step. Mix and aliquot 90-120 µL to a polystyrene sonication tube.
- Add an equal volume of resuspension buffer containing 2x detergents (150 mM NaCl, 50 mM HEPES-KOH [pH 7.5], 1 mM EDTA, 0.2% sodium deoxycholate, 0.7% sarkosyl).
- Sonicate in a waterbath sonicator at 50% power for 7 min (20 s on/40 s off) at 4 °C. Gently mix by pipetting. Repeat the sonication for an additional 7 min.
- Transfer the sonicated lysate to a 1.5 mL tube. Add 0.5 volumes of resuspension buffer without detergents (150 mM NaCl, 50 mM HEPES-KOH [pH 7.5], 1 mM EDTA) (e.g., add 100 µL of buffer to 200 µL of sample).
- Centrifuge at 13,000 x g for 15 min at 4 °C. Keep the lysate supernatant and transfer to a new tube.
- Optionally, perform a protein assay to determine the concentrations of each lysate. This step may be useful for large sample quantities or inter-assay comparisons. However, standardizing the number of animals as described above works well for the sample quantities described here, since there is better correlation with the yield of chromatin/DNA as determined by qPCR.
7. Immunoprecipitation
NOTE: Scale the amount of antibody and magnetic beads to the volume and concentration of lysate.
- Divide the lysate supernatant into four portions: three equal volumes for each IP and 10% of one IP's volume as the Input (e.g., 100 µL of IP #1, 100 µL of IP #2, 100 µL of IP #3, 10 µL of Input). Transfer the IP lysate to a 200 µL PCR tube and store the Input lysate at -20 °C in a 1.5 mL tube.
- Add 0.5 µg of anti-H3K9me3 antibody, anti-histone H3 antibody, or IgG to the appropriate IP sample. Incubate at 4 °C overnight with rotation.
- The following day, aliquot 9 µL of protein G-coated magnetic beads per IP to a single 1.5 mL tube. Wash the beads twice with 1 mL of FA-150 (150 mM NaCl, 50 mM HEPES-KOH [pH 7.5], 1 mM EDTA, 1% TX-100, 0.1% sodium deoxycholate).
- Resuspend the magnetic beads in FA-150 buffer to the original volume taken from the stock in step 7.3 above. Add 7.5 µL to each IP. Incubate at 4 °C for 2 h with rotation.
8. Washes and elution
NOTE: To ensure that the magnetic beads do not dry, add each wash or elution buffer quickly after aspirating the previous wash.
- Use 0.2-1 mL of the following buffer solutions each time to wash the beads. Collect the beads on a magnetic stand to aspirate the washes. Incubate each wash at 4 °C for 5 min with rotation. Follow the order as below.
- Wash twice with FA-150.
- Wash once with FA-1M (FA-150 with 1 M NaCl).
- Wash once with FA-0.5M (FA-150 with 0.5 M NaCl). Transfer to a new PCR tube.
- Wash once with TE-LiCl (250 mM LiCl, 10 mM Tris-Cl [pH 8.0], 1 mM EDTA, 1% IGEPAL CA-630, 1% sodium deoxycholate).
- Wash twice with TE+ (50 mM NaCl, 10 mM Tris-Cl [pH 8.0], 1 mM EDTA, 0.005% IGEPAL CA-630).
NOTE: Continue sample processing at room temperature unless otherwise indicated.
- Aspirate the last wash buffer. Resuspend the magnetic beads with 50 µL of ChIP elution buffer (200 mM NaCl, 10 mM Tris-Cl [pH 8.0], 1 mM EDTA, 1% sodium dodecyl sulfate [SDS]) and transfer to a 1.5 mL tube.
- Elute at 65 °C for 15 min in a thermomixer with mixing at 1,000 rpm for 5 s each minute.
- Collect the beads on a magnetic stand and transfer the supernatant to a new tube.
- Repeat the elution with another 50 µL of ChIP elution buffer. Pool the supernatants for a total of 100 µL. Proceed to reverse crosslinking and DNA elution, as detailed below.
9. Reverse crosslinking and DNA elution
- Thaw the Input lysate sample. Top up to 100 µL with ChIP elution buffer.
- Add 16.5 µg of RNase A to each IP and Input sample. Incubate at 37 °C for 1 h.
- Add 40 µg of Proteinase K and incubate at 55 °C for 2 h. Then, incubate at 65 °C overnight.
- Cool the samples to room temperature and purify the DNA with a spin-column kit.
10. qPCR reaction setup and run
NOTE: The primer, reaction setup, and thermocycler parameters should be modified to match the manufacturer's recommendations for the qPCR reaction mix in use.
- Prepare primer sets targeting the GFP RNAi reporter and H3K9me3 positive and negative enrichment control regions. The primer melting temperature is 60 °C. See Table of Materials for primer sequences.
- For the Input DNA, make four fourfold serial dilutions (e.g., 1:5, 1:20, 1:80, 1:320).
NOTE: The IP DNA may be used directly or diluted (e.g., 1:2 or 1:3) to allow for more reactions. The suitability of the dilution must be determined empirically, since the qPCR performance may be affected. - Organize all the reactions corresponding to one primer set on one PCR plate. Set up technical duplicate reactions for each Input DNA dilution and each IP DNA sample. Each 10 µL reaction contains 1.5 µL of Input or IP DNA (or elution buffer as a control), qPCR master mix (1x final concentration), forward primer, and reverse primer (400 nM final concentration for each primer).
- On a real-time thermocycler, run the following program: initial denaturation: 95 °C for 4 min; amplification and fluorescence detection: 40 cycles of 95 °C for 10 s and 60 °C for 30 s with a plate read; final extension: 60 °C for 5 min; melt curve: from 60 °C to 90 °C in 0.5 °C increments, 5 s per step.
11. Determining amplification efficiency and verifying product specificity
- For each set of Input DNA dilutions, plot the four data points with [log10(1/dilution)] on the x-axis and [Input Cq] on the y-axis. Determine the slope of the line of best fit.
- Calculate the amplification efficiency. Ideal primer sets should consistently display 95%-100% efficiency.
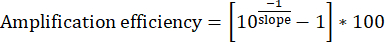
- Check that all the reactions have one sharp melt-curve peak and that reactions with the same primer set have the same melting temperature. Multiple peaks or different melting temperatures may indicate non-specific amplification.
- Optionally, run reactions on a standard 2% agarose/TAE gel at room temperature to verify product band size.
12. Calculating the percentage of Input
- Calculate the Input dilution factor. Since 10% of IP lysate volume was saved as Input, the dilution factor for the 1:5 dilution of Input DNA is 50.
- Determine the IP dilution factor. If the IP DNA is not diluted before qPCR, then the dilution factor is 1.
- Calculate the Cq difference between the IP and Input adjusted for dilution factors.
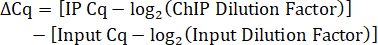
- Calculate the percentage of Input.
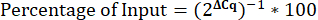
Animals carrying the germline-expressed GFP-histone H2B [mex-5p::gfp-h2b::tbb-2 3'UTR]7 reporter (Figure 2A) were exposed to GFP RNAi or control RNAi by feeding, and passaged as described in the protocol and Figure 1. Nuclear GFP signal in the germline was manually scored using a fluorescence dissecting microscope for a sample of the population at each generation. Silencing of the transgene was fully penetrant in the scored P0 and F1 animals treated with GFP RNAi (Figure 2B). In the F2 generation, the proportion of the population exhibiting inheritance of GFP silencing was approximately 50%. By the F5 generation, the majority of the population did not show inheritance of silencing, and by the F10 generation, no inheritance was detected, as all the animals expressed GFP.
To determine the change in histone H3K9me3 enrichment corresponding to RNAi-induced silencing, ChIP-qPCR was performed on F1 generation animals following either GFP RNAi or control RNAi treatment. As expected, the GFP RNAi-treated population displayed higher histone H3K9me3 levels at the GFP target and at the 1.3 kb downstream region compared to control RNAi-treated animals (Figure 2C). The specificity of the histone H3K9me3 ChIP is supported by the enrichment at a positive control locus (clec-18) known to be enriched in this mark, but not at a nearby negative control locus (hrp-2). Histone H3 enrichment and near-background enrichment in the IgG control immunoprecipitations were also detected at all qPCR loci, as expected. When ChIP enrichment at the reporter is normalized to the clec-18 positive control locus, higher histone H3K9me3 enrichment upon GFP RNAi is shown, whereas histone H3 enrichment is similar between the control and GFP RNAi treatments (Figure 2D). Since GFP RNAi is not expected to affect histone H3K9me3 or total histone H3 occupancy at the clec-18 locus, this normalization mitigates against technical variation, such as differences in ChIP efficiency between the GFP RNAi and control RNAi samples. The fold-change of histone H3K9me3 and histone H3 levels between RNAi treatments shows GFP reporter-specific histone H3K9me3 enrichment, independent of histone occupancy, upon GFP RNAi-induced silencing (Figure 2E).
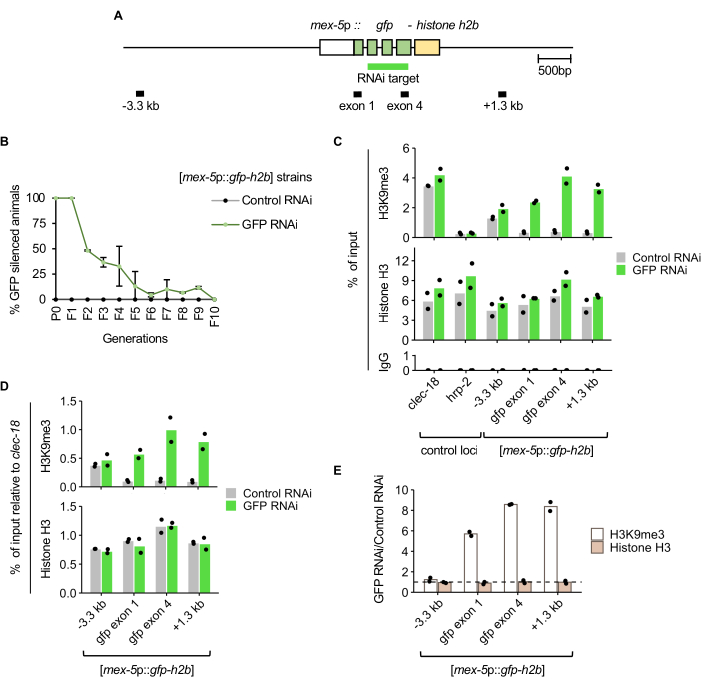
Figure 2: GFP RNAi-induced silencing corresponds to elevated H3K9me3 enrichment at the RNAi target. (A) Diagram of the germline-expressed GFP RNAi reporter mjIs134[mex-5p::gfp-h2b::tbb-2 3'UTR] with qPCR amplicon regions labelled. (B) GFP expression scored across generations after RNAi treatments at 21 °C. Error bars represent the standard deviation from two biological replicates. (C) ChIP-qPCR of H3K9me3, histone H3, and IgG control in F1 young adults from two biological replicates. clec-18 and hrp-2 are the positive and negative control loci for H3K9me3 enrichment, respectively. (D) H3K9me3 and histone H3 enrichment at the GFP RNAi reporter normalized to the clec-18 positive control locus. (E) Change in H3K9me3 and histone H3 enrichment between GFP RNAi- and control RNAi-treated animals, with normalization to clec-18. Dotted line represents a fold-change of 1. Please click here to view a larger version of this figure.
