We describe here a protocol for the differentiation and mechano-stimulation of endothelial cells derived from hiPSCs that allows the study of their response to mechanical cues (Figure 1). This protocol results in the production of functionally mechanosensitive endothelial cells. We provide here representative results and describe the expected phenotype to assess how the cells respond to the cytokine stimulation during the differentiation.
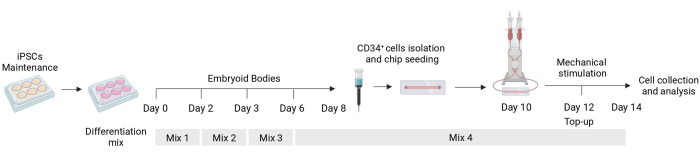
Figure 1: Schematic of the differentiation and mechanical stimulation protocol. Schematic of the differentiation protocol showing the timing of the different mixes of cytokines, the CD34+ cell isolation, fluidic chip seeding, and final analysis of the mechanically stimulated cells. Please click here to view a larger version of this figure.
Culture of hiPSCs
It is important to start the protocol from hiPSCs that are growing correctly in self-renewal conditions. A good indication of the quality of the culture is the speed of their growth. After thawing, the cells might need 2-3 weeks to reach the correct phase of growth that will ensure good differentiation. When the cells can be passaged twice a week at the ratio of 1:6 reaching almost full confluency, this is the time that they are ready to be differentiated on the same day they need to be passaged.
Differentiation of hiPSCs into endothelial cells
The first step of the differentiation, consisting of the formation of embryoid bodies (EBs), is cell line-dependent and may need some optimization for the specific cell line in use. The dissociation described in protocol steps 1.3.2.2-1.3.2.4 can be modified by either reducing or extending the incubation with the dissociation reagent and the subsequent dissociation with the Pasteur pipette. Furthermore, other dissociation reagents can be used for this step in addition to the physical dissociation of the colonies with a cutting tool or a P100 pipette tip. EBs of good quality show a defined edge by day 2 of the differentiation and appear clear and bright when observed using a microscope; darker areas might indicate cell death within the EBs (Figure 2).
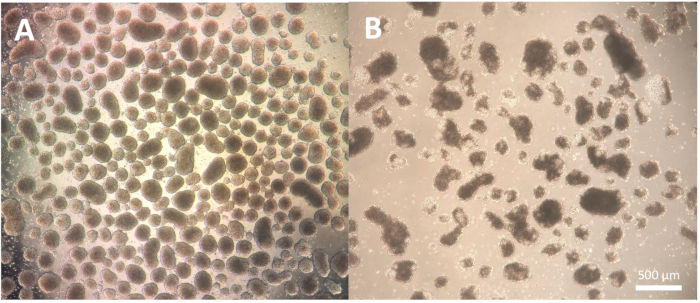
Figure 2: Embryoid bodies morphology. (A) Day 2 embryoid bodies showing well-defined outer edges and consistent size. (B) Day 2 embryoid bodies of poor quality showing extensive cell death leading to disaggregation of the structure. Scale bar = 500 µm. Please click here to view a larger version of this figure.
At day 2, the addition of CHIR99021 to the EBs inhibits the GSK-3 protein resulting in the activation of the Wnt pathway. Different cell lines have different responses to CHIR treatment, and this should be tested by quantifying the number of CD34+ cells obtained at day 8 by using different concentrations (Figure 3).
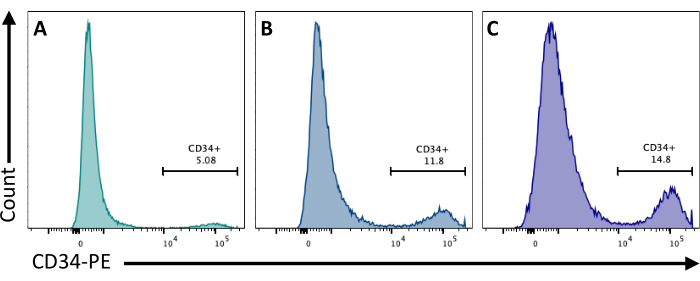
Figure 3: Endothelial cell differentiation with different CHIR treatments. Endothelial cell commitment quantified by flow cytometry at day 8 of differentiation by CD34 membrane expression, following CHIR treatment at day 2 at (A) 3 µM, (B) 5 µM, and (C) 7 µM. Flow cytometry data were obtained using five-laser cytometers and dedicated software (see Table of Materials). Please click here to view a larger version of this figure.
CD34+ cell isolation
It is important to validate that the CD34+ enrichment using the magnetic beads provides at least 80% CD34+ after elution of the column. To ensure sufficient purity, an aliquot of cells obtained from the magnetic isolation can be analyzed by flow cytometry making sure to use a different antibody clone than the one used for the magnetic enrichment. Here, the 4H11 clone was used and ~85% purity was achieved post enrichment (Figure 4).
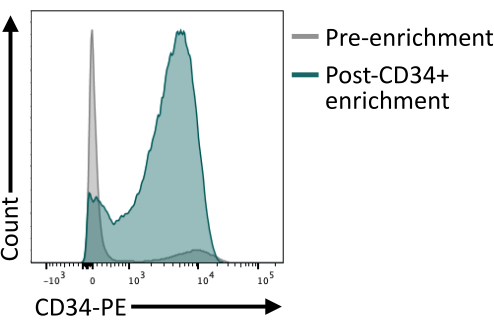
Figure 4: Membrane expression of CD34 before and after enrichment by magnetic sorting. Day 8 dissociated embryoid bodies (grey) and cells after magnetic enrichment (green) were stained for CD34 expression and analyzed by flow cytometry, showing successful enrichment post sorting. Flow cytometry data were obtained using five-laser cytometers and dedicated software (see Table of Materials). Please click here to view a larger version of this figure.
Seeding cells into the fluidic channel
When seeding the cells in the fluidic channel, it is crucial to track the adhesion and proliferation of the endothelial cells. After seeding, the cells take ~5 h to fully adhere to the channel (Figure 5A). An alternative coating solution may also be tested to improve adhesion at this stage. To confirm that the tested cells are mechanosensitive and thus, able to respond to mechanical stimulation, the cell orientation can be tested over time. Cells before the stimulation show random orientation (Figure 5A and Figure 5C) and they reorient parallel to the direction of the flow (Figure 5B,C). The protocol described here allows for the collection of the cells from the channel to perform downstream analysis, for example, flow cytometry, for the study of their membrane immunophenotype, providing the endothelial identity of the stimulated cells (Figure 5D,E).
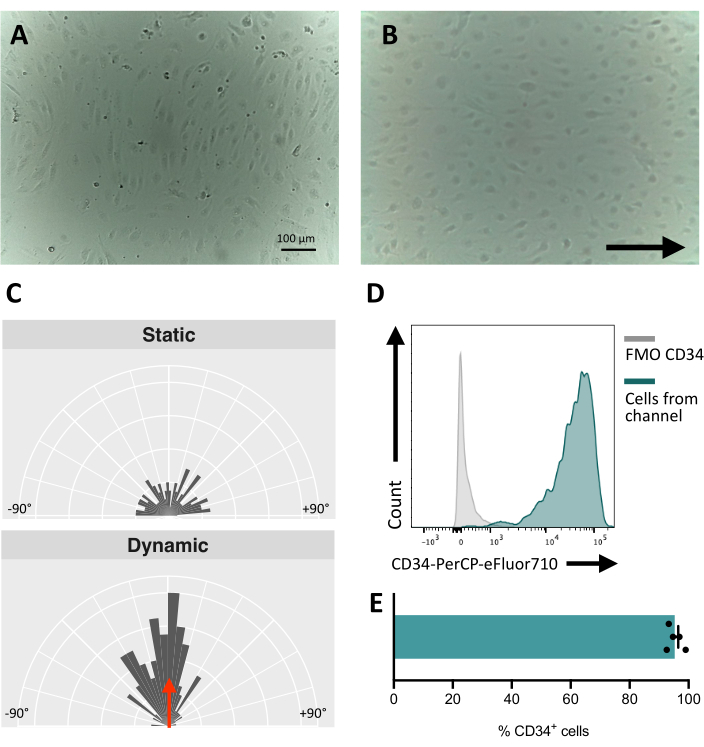
Figure 5: Mechanoresponsiveness of hiPSCs-derived endothelial cells. (A) Confluent layer of isolated CD34+ cells 48 h post seeding. (B) Reoriented layer of endothelial cells 3 days under dynamic culture. (C) Orientation analysis of the endothelial cells after 5 days of dynamic culture. (D) CD34 expression profile of cells cultured under flow for 5 days. (E) Percentage of CD34+ cells of cell population retrieved from the fluidic channel. Images were taken using an inverted in-incubator microscope; flow cytometry data were obtained using five-laser cytometers and dedicated software (see Table of Materials). Scale bars = 100 µm (A,B). Please click here to view a larger version of this figure.
| Reagents | Stock concentration | Volume added | Final concentration |
| Iscove’s Modified Dulbecco’s Medium (IMDM) | – | 333 mL | – |
| Ham’s F-12 Nutrient mixture (F-12) | – | 167 mL | - |
| N-2 supplement (100x) | 100 x | 5 mL | 1x |
| B-27 supplement (50x) | 50 x | 10 mL | 1x |
| Ascorbic acid | 10 mg/mL | 1.25 mL | 25 µg/mL |
| α-Monothioglycerol (MTG) | 11.5 M | 19.5 µL | 448.5 µM |
| Human Serum Albumin | 100 mg/mL | 2.5 mL | 0.5 mg/mL |
| Holo-Transferrin | 100 mg/mL | 0.75 mL | 150 µg/mL |
Table 1: Composition and recipe for 500 mL of Serum-free Differentiation (SFD) medium.
| Days of differentiation | Cytokine Mix | Cytokine | Final concentration |
| Day 0 – 2 | Mix 1 | BMP4 | 20 ng/mL |
| Day 2 | Mix 2 | CHIR99021 | 7 μM |
| From day 3 onward | Mix 3 and 4 | VEGF | 15 ng/mL |
| bFGF | 5 ng/mL | ||
| From day 6 onward | Mix 4 | IL6 | 10 ng/mL |
| FLT3L | 10 ng/mL | ||
| IGF1 | 25 ng/mL | ||
| IL11 | 5 ng/mL | ||
| SCF | 50 ng/mL | ||
| EPO | 3 U/mL | ||
| TPO | 30 ng/mL | ||
| IL3 | 30 ng/mL |
Table 2: Mixes of cytokines used for endothelial cell differentiation, days in which they are added to the SFD medium, and final concentration.
| Shear Stress (dyn/cm2) | Time (h) |
| 0.5 | 1 |
| 1 | 1 |
| 1.5 | 1 |
| 2 | 1 |
| 2.5 | 1 |
| 3 | 1 |
| 3.5 | 1 |
| 4 | 1 |
| 4.5 | 1 |
| 5 | Until end of the experiment |
Table 3: Shear stress values for the dynamic culture and length of their application.
Supplementary Figure S1: Geometry and dimensions of the chip and tubing used for this protocol. Please click here to download this File.
Supplementary Figure S2: Step-by-step guide for the software controlling the air pump with a description of each step. Please click here to download this File.
Supplementary Figure S3: Guide for the orientation analysis using FIJI showing the drawing of the cell shape, elliptic fitting, and final measurement. Please click here to download this File.
Supplementary Table S1: Unit size, resuspension volume, and stock concentrations for cytokines used in differentiation protocol. Please click here to download this File.
