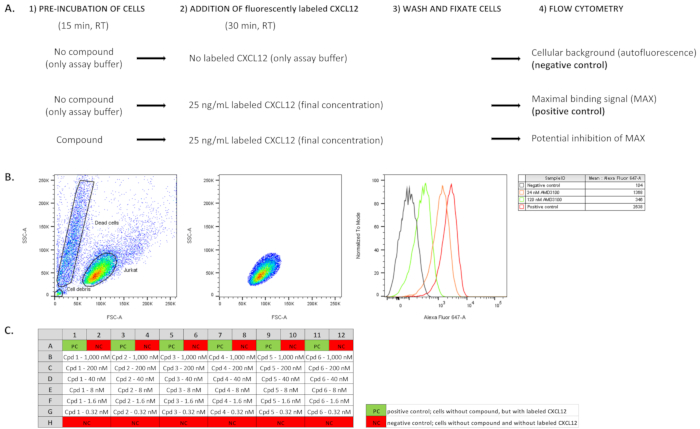
Figure 1: Overview of the workflow and illustration of the type of data obtained. (A). The main steps of the protocol are schematized for the negative and positive control samples, and compound-treated samples. Samples without compound pre-incubation and without addition of fluorescently labeled chemokine (CXCL12AF647) are included to determine the background (auto)fluorescence (negative control samples). Samples without compound pre-incubation, but with addition of a fixed amount of CXCL12AF647 are used to determine the maximal binding signal in the assay (positive control samples). In compound-treated samples, cells are pre-incubated with compound before addition of a fixed amount of CXCL12AF647. (B). The mean fluorescent binding signal is determined by flow cytometry analysis of Jurkat cells. From all events (i.e., Jurkat cells) analyzed (left panel) only the fluorescent signal from a gated subpopulation of cells is used for further analysis (middle panel). Pre-incubation of cells with small molecules (e.g., AMD3100) inhibits the binding of the fluorescently labeled receptor ligand and this will reduce the maximal binding signal (right panel, shown in a histogram representation). (C). A possible plate layout when performing the competitive binding assay in a 96-well plate format. In this case, six different compounds (cpd 1- 6) are tested in a 1/5 serial dilution series (in duplicate) ranging from 1,000 nM down to 0.32 nM.
