1) Cell Culture
- Select cell types expressing the gene of interest.
- Select cell lines heterozygous both for DNA risk variants and transcribed coding polymorphism within the gene of interest (Figure 1).
- Culture the selected cell lines according to the supplier’s specifications and prepare 2 x 10 cm petri dishes for DNA and RNA extraction respectively.
- When cells reach 80% confluence start the procedure to harvests cells and isolate DNA and RNA either following the procedure described below or using commercially available kits.
2) DNA Extraction
- Remove media from the dish, wash with PBS and add to the dish 500 μL of a lysis solution (0.6% SDS, 100 mM NaCl, 50 mM Tris, 20 mM EDTA and 50 μg/mL RNase A).
- Rock the plate gently for 20 minutes at room temperature.
- Scrape the cell lysate into a microcentrifuge tube and incubate at 37 °C overnight.
- Add proteinase K to a final concentration of 100 μg/mL and incubate at 37 °C overnight.
- Extract the DNA with an equal volume of phenol-chloroform pH 8, repeat, then extract with an equal volume of chloroform/isoamyl alcohol.
- Precipitate the DNA with 1/10th volume 3 M NaCl and 2.5 volumes of 100% ethanol.
- Leave on ice for 5 min and then spin at 13,000 rpm for 30 min at 4 °C.
- Wash the DNA with 70% ethanol, allow to air-dry and resuspend in 200 μL of TE.
- Incubate at 65 °C for 10 min and then leave for 2 days at room temperature.
- Store at 4 °C or long term at -20 °C.
3) RNA Extraction
- Remove media from the dish, wash with PBS, add 1 mL of TRIzol and incubate for 10 min at room temperature.
- Scrape cells, pipette a few times, transfer in a microcentrifuge tube and incubate for 5 min.
- Spin for 10 min at 10,000 rpm at 4°C.
- Transfer the supernatant in a fresh tube and add 200 μL of chloroform.
- Shake and spin for 10 min at 10,000 rpm at 4°C.
- Transfer the aqueous phase in a fresh tube and add an equal volume of 70% ethanol.
- Load the solution to 1 or 2 (depending on the volume) RNeasy column and spin for 30 sec at maximum speed.
- From here follow instruction of the RNeasy kit (Qiagen).
4) Nucleic Acid Sample Preparation
- Quantify DNA and RNA concentration using a NanoDrop Spectrophotometer (Thermo Scientific).
- Prepare cDNA from 500-1000 ng of RNA using random decamers and SuperScript III Reverse Transcriptase (Invitrogen) according to the manufacturer’s instructions.
5) PCR and Primer Extension Assay
- Design two pairs of PCR primers for each DNA marker, one pair for genomic DNA amplification and the other pair for cDNA amplification (Fig 2). Place forward and reverse primer for cDNA amplification in different exons to avoid genomic contamination. Design primer to obtain a PCR product in the range of 100-500 bp length.
- Prepare a 10 μL PCR reaction including 0.5 U Immolase Taq (Bioline) 0.8 mM dNTPs, 2 mM MgCl2, 0.2 M each primer and 20 ng of either cDNA or DNA. Amplify each sample in four independent reactions.
- Run the PCR reaction under the conditions optimized for each primer pair keeping cycle number in linear phase of amplification.
- Remove PCR primer and dNTPs excess by incubating the PCR product with exonuclease I and Shrimp Alkaline Phosphatase (SAP) at 37 °C for 20 minutes.
- Spot each PCR product twice on a 384-well plate, so that 8 measurements will be available for each sample.
- Design the extension primer with the Assay Design (Sequenom) software so that the same primer can be used for both genomic and cDNA PCR products. Specify primer length in the range of 14-18 bp and extension mix including combinations of 3ddNTPs (Fig 2).
- Carry out the primer extension reaction and MALDI-TOF/MS analysis using the MassARRAY system (Sequenom) according to the manufacturer’s instruction.
- A typical primer extension reaction include a starting step at 94 °C for 2 min, followed by 40 cycles of 94 °C for 5 s, 52 °C for 5 s and 72 °C for 5 s.
- The primer extension products are cleaned with SpectroCLEAN resin and transferred onto microarray (chip) by SpectroPOINT nanolitre dispenser.
- The extended oligonucleotides are quantified by MALDI-TOF using a SpectroREADER mass spectrometer.
6) Statistical Analysis
- Check MALDI-TOF/MS results with the MassARRAY Typer software for general quality of the experiment.
- Extract the Allelotyping report which includes the peak area data.
- For each individual spectrum calculate the ratio between the peak area of allele 1 and allele 2 (peak area ratio).
- For each cDNA sample derive a peak area ratio mean and standard deviation, using the corresponding function in Excel, across the 8 measurements.
- Derive the same mean peak area ratio across the 8 measurments for genomic DNA.
- Divide the cDNA mean ratio by the genomic mean ratio to assess the deviation to the expected 1:1 ratio.
7) Experimental Controls
- To rule out experimental artifacts, repeat the experiment at least three times.
- Whenever possible, design assays for additional DNA markers.
- Design multiple PCR primer pairs and extension primers annealing to both forward and reverse strands.
- Whenever possible test as negative controls cell lines that are heterozygous for DNA markers but do not carry the DNA risk variants associated with the phenotype (Fig. 1 and Fig. 3)
8) Representative Results
An example of allele-specific expression analysis is shown in Figure 3 with examples of mass spectrometry traces. The figure shows the results from the analysis of 4 allele-specific primer extension products carried out on 4 different templates. The top graphs represent the results obtained from the analysis of genomic DNA of two different cell lines, which are both heterozygous for a transcribed coding polymorphism. However, only cell line A is heterozygous for the risk DNA variant (Figure 1). The lower graphs represent the results obtained from the analysis of the cDNA generated from the same cell lines. As a result of experimental artifact the first peak is always higher than the second peak even in the genomic analysis. Therefore, the results from the genomic analysis are used to normalize the cDNA data. After normalization, the allele ratio is significantly different from 1 in cell line A (where the second peak appears lower compared to the other spectra), while it is very close to 1 in cell line B. The data suggest that cell line A carries a DNA variant, in phase with the allele measured by the second peak, which reduces the expression of the gene under analysis. Cell line B provides a very convenient negative control.

Figure 1. Cell line selection strategy. Cell lines A and B are heterozygous for a transcribed coding marker (triangle) but only cell line A is heterozygous for risk DNA variant (circle). The blue allele of the risk variant is associated (either with a direct effect or because in close correlation with the actual functional variant) with a lower level of transcription.
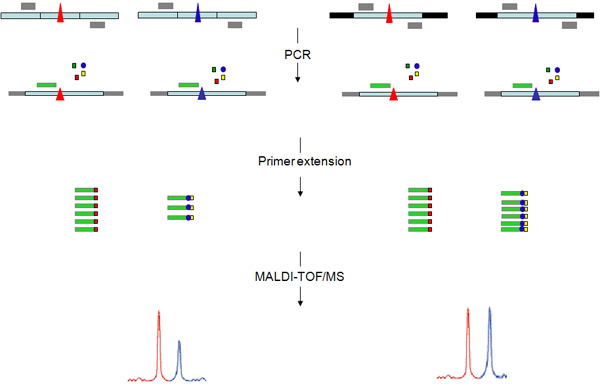
Figure 2. Allele-specific primer extension assay. This figure show the work-flow of the experimental procedure carried for both cDNA (left) and genomic DNA (right) templates. PCR primers (grey rectangles) are designed to amplify a heterozygous coding polymorphism (triangle). To avoid genomic contamination PCR primers anneal sequences placed in different exons (light blue bars) when amplification is carried out on cDNA template. The PCR product is then used as template for a primer extension reaction carried out with an extension primer, annealing one base next to the polymorphism, and the appropriate mix of three dideoxynucleotide (ddNTPs) terminators (squares) and one deoxynucleotide (circle) to extend the primer of one or two basis respectively. The resulting primer extension products have different masses which allow separation and quantification by MALDI-TOF mass spectrometry. The quantity of product corresponding to the blue allele of the DNA marker is relatively lower to the red allele.
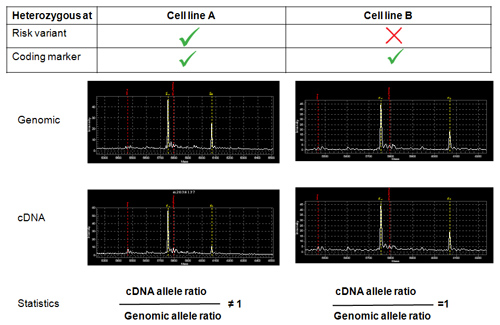
Figure 3. Results of an allele-specific expression assay. The figure show the mass spectrometry results from the analysis carried out on the genomic DNA and the cDNA of cell line A and cell line B (Figure 1).
