Protocol A: Stable isotopic profiling of intermediary metabolism during C. elegans development on NGM plates.
*Note: Stable isotope enrichment can be successfully measured in young adult nematode populations following isotope exposure (with either universally-labeled 13C glucose or 1,6-13C2-glucose) beginning either in early development or in early adulthood. This protocol facilitates concurrent monitoring of animal health, development, and growth on standard nematode growth media (NGM) plates, which is not as easily achieved in liquid culture.
- Prepare 100 mm Petri dishes with 10 mL of nematode growth media (NGM), per standard technique. 1
- Spread NGM plates with OP50 E. coli 1 and allow to dry overnight.
- Add 500 μL of 200 mM 1,6-13C2 glucose (to achieve a final concentration of 10 mM on NGM plate) uniformly to plate. Allow to dry overnight.
- Bleach gravid adult worms 1. Plate recovered eggs on NGM plates without bacteria or 1,6-13C2 glucose. Incubate at 20°C overnight.
- The next day, transfer hatched, L1-arrested larvae onto NGM plates previously spread with 1,6-13C2-glucose and OP50 E. coli (per step #2, above). Grow worms to young adult stage (48-72 hours, depending on strain), taking care worms do not starve.
- Collect synchronous, first-day young adult worms from plates with 3-4 mL of S. Basal.
- Wash worms in 50 mL of S. Basal in 50 mL Falcon tubes. Allow worms to pellet by gravity for 5 minutes. Remove supernatant to ~5 mL. Repeat wash twice more.
- Estimate worm number by counting three 100 μL aliquots2. Adjust worm concentration to 1000 worms/mL of S. basal.
- Pipette 1 mL (1000 worms) into a 1.5 mL plastic centrifuge tube.
- Add 69 μL of 60% PCA containing internal standard (ε-aminocaproic acid) to a final concentration of 4% PCA and 200 μmol of internal standard.
- Let worms settle by gravity x 5 minutes. Remove and save the supernatant in another tube.
- Grind worm samples using plastic homogenizer (Kontes Pellet Pestle) and motorized drill (Kontes Pellet Pestle Cordless Motor) for 15 seconds. Visually confirm worm disruption by light microscopy. Repeat if necessary.
- Transfer the supernatant from step #11 to combine with homogenate from step #12.
- Centrifuge sample at 2250 rpm (1300 x g) for 5 minutes.
- Transfer supernatant to fresh 7 mL glass tube. Save pellet for protein concentration assay, as detailed in step #20, below.
- pH samples to 7-8 (neutral) range using 4N KOH.
- Centrifuge neutralized samples in 7 mL glass tube at 2250 rpm (1300 x g) for 5 minutes to remove salts.
- Transfer supernatant to fresh 7 mL glass tube.
- Prepare neutralized samples for metabolite quantitation by:
- High Performance Liquid Chromatography (HPLC): Separate 50 μL of neutralized sample for direct injection into HPLC. Free amino acid quantitation is performed by HPLC (Varian) using pre-column derivatization with o-phthalaldehyde and fluorescent detection, as previously described3-4.
- Gas Chromatography/Mass Spectrometry (GC/MS): Proceed with extraction of remaining neutralized samples using ion exchange resin (Bio-Rad) for GC/MS determination of isotopic enrichment in amino acids and organic acids, as detailed in PROTOCOL D.
- Add 1 mL of 1N NaOH to remaining protein pellet (from step #15) in 7 mL glass tube.
- Incubate NaOH-treated protein sample at room temperature while rotating (Labnet* LabRoller Rotator) overnight until dissolved.
- Use 75-100 μL of NaOH-treated protein to determine protein concentration by standard methods (DC Protein Assay, Bio-rad).
Protocol B. Stable isotopic profiling of intermediary metabolism in C. elegans young adults in liquid culture.
*NOTE: Pharmacologic effects on intermediary metabolic flux in adult nematodes can be studied following isotopic exposure beginning on the first day of egg laying either on NGM plates (see Protocol A, above) or in liquid culture. We prefer the latter approach to maximize cost efficiency of stable isotope and pharmacologic agent utilization.
- Dilute synchronous, first day egg-laying young adults in S. basal containing 0.0005% cholesterol (obtained by adding 0.5 mL of 1% cholesterol (dissolved in 95% Ethanol) to 1L of S. Basal) to 2000 animals per 500 μL.
- Set up ~ 4 mL total volume culture(s) in 25 mL Erlenmeyer flasks, as follows:
TREATMENT: NONE “DRUG A” S. Basal with cholesterol 3020 μL 3020 uL – “Drug A” volume Stable Isotope (1,6-13C2-glucose) 80 μL 80 μL K12 E. coli (OD660 2.5 to 3) 400 μL 400 μL Young Adult Worms (2,000) 500 μL 500 μL Drug A (desired concentration) – As desired - Cover flasks with foil. Shake flasks for 24 hours at 220 rpm in 20°C incubated platform shaker.
- Make sure flasks are not completely sealed, as oxygen is required for intermediary metabolic flux and for resulting labeling of endogenous nematode metabolites.
- Wash worms by adding 4 mL culture volume to 46 mL of S. basal in a 50 mL conical plastic tube. Allow worms to pellet by gravity for 5 minutes. Vacuum suction supernatant down to ~ 5 mL remains. Repeat wash by refilling tube twice more to 50 mL with S. basal.
- Concentrate washed worms to 1000 worms/mL in a 1.5 mL plastic centrifuge tube.
- Add 69 μL of 60% perchloric acid (PCA) and 2 μL of 10 μM internal standard (ε-aminocaproic acid) to achieve a final concentration of 200 μmol internal standard and 4% PCA.
- Prepare samples as per steps #11-22 in PROTOCOL A.
PROTOCOL C-1. Monitoring isotopic utilization in adult worms on plates by tracing labeled carbon in atmospheric and dissolved carbon dioxide.
*NOTE: Whereas PROTOCOLS A and B detail methods to monitor isotope incorporation into free metabolites within worms over 2-3 days of development or 1 day of adult life, respectively, gross confirmation of the success and kinetics of isotopic incorporation over a short time course (i.e., minutes to hours) can be achieved by monitoring label in atmospheric carbon dioxide (CO2) released from worms or contained in dissolved carbon dioxide within worm extract. Live or killed bacteria can be fed to worms when grown on plates (PROTOCOL C-1). Bacteria is not necessary for short-term isotope exposure of worms in liquid culture (PROTOCOL C-2).
- Prepare 100 mm Petri dishes with 10 mL of NGM agar. Spread NGM plates with either live or UV-killed OP50 E. coli (as demonstrated in Figure 1). Allow plates to dry overnight.
- Add 500 μL of 200 mM 1,6-labeled-13C2-glucose to OP50 spread NGM plates (for final isotope concentration of 10 mM on NGM plate). Allow plates to dry overnight.
- Obtain synchronous, first day egg-laying, young adult worms grown on NGM agar plates spread only with OP50 E. coli.
- Transfer 1000 young adult worms to experimental NGM agar plates containing stable isotope and E. coli (prepared as per steps #1-2, above).
- Place experimental NGM plate (without cover) in custom glass chamber fitted with well-sealed 3-way stopcock to allow for precise atmosphere sampling and replacement. Immediately seal chambers with optically transparent glass disc. Cover seam where glass disc sits on plate with high vacuum grease to seal. Record time as “Time 0”.
- Sample 10 mL of atmosphere from glass chamber by 3-way stopcock with a 20 mL syringe at 30, 60, 90 and 120 minutes. Transfer each atmospheric sample to a pre-prepared 10 mL rubber stopper topped glass tube containing 1 mL of 1 mM NaHCO3 in 0.1 N NaOH that has been vacuum sealed.
- Inject 10 mL of air back into each glass chamber through 3-way stopcock using a 20 mL syringe. Close stopcock to chamber after sampling/reinjecting atmosphere and before resuming incubation between collection time points.
- Inject 100 μL of 20% phosphoric acid through stopper into 10 mL rubber stopper topped glass tube containing atmospheric CO2 sample.
- Remove 2 mL of atmosphere and inject it into 12 mL blue-top auto-sampler tubes pre-filled with helium for atmospheric CO2 measurement.
- Analyze “atmospheric CO2 samples” by Gas-Ratio Mass Spectrometer (ThermoQuest Finnigan Delta Plus).
- Open glass chambers following two hour isotope incubation period and collection of all atmospheric samples.
- Wash worms off of NGM plates using 3 mL of S. basal.
- Wash worms in 50 mL S. basal in 50 mL Falcon tubes. Repeat wash two more times.
- Concentrate worms to ~1000 worms/mL in 7 mL round-bottom glass tube(s).
- Vacuum seal glass tube with rubber stopper.
- Add 100 μL of 20% phosphoric acid through rubber stopper with syringe to release dissolved CO2.
- Remove 2 mL of atmosphere and inject into 12 mL blue-top auto-sampler tubes pre-filled with helium for dissolved CO2 measurement.
- Analyze “dissolved CO2 samples” by Gas-Ratio Mass Spectrometer (ThermoQuest Finnigan Delta Plus).
Alternative Protocol (C-2): Monitoring isotopic utilization in adult worms in liquid culture by tracing labeled carbon in atmospheric and dissolved carbon dioxide.
- Obtain synchronous, first day egg-laying, young adult worms grown on NGM agar plates spread with OP50 E. coli.
- Wash worms in 50 mL of S. basal in 50 mL Falcon tubes. Allow worms to pellet by gravity for 5 minutes. Remove supernatant to ~5 mL. Repeat washes twice more.
- Transfer 1000 young adult worms in 1 mL S. basal to 10 mL round-bottom glass tube. Add 10.1 μl of 1 M stock of universally-labeled 13C-glucose to 1 mL of worms to achieve final concentration of 10 mM.
- Oxygenate the round-bottom glass tube containing worms and isotope by exchanging atmosphere with flowing 100% oxygen in open tube for 2 minutes. Immediately close tube with rubber stopper.
- Shake experimental tubes in 20°C incubated platform shaker at 220 rpm. Record starting time as “Time 0”.
- Sample 5 mL of atmosphere from 10 mL round-bottom glass tube using a 10 mL syringe and 25 gauge needle through the rubber stopper at 30, 60, 90 and 120 minutes.
- Transfer each atmospheric sample to a pre-prepared, rubber stopper topped 10 mL glass tube containing 1 mL of 1 mM NaHCO3 in 0.1 N NaOH that has been vacuum sealed.
- Inject 5 mL of air back into each experimental round-bottom glass tube containing worms and isotope through rubber stopper using a 10 mL syringe and 25 gauge needle.
- Resume incubation of experimental 10 mL round-bottom glass tubes containing worms and isotope by shaking at 220 rpm between collection time points.
- Inject 100 μL of 20% phosphoric acid through stopper into atmosphere containing rubber stopper topped 10 mL glass tube.
- Remove 2 mL of atmosphere and inject into 12 mL blue-top auto-sampler tubes pre-filled with helium for atmospheric CO2 measurement.
- Analyze “atmospheric CO2 samples” by Gas-Ratio Mass Spectrometer (ThermoQuest Finnigan Delta Plus).
- At end of experimental period, wash worms in 50 mL S. basal in 50 mL Falcon tube. Allow worm pellet to form by gravity. Vacuum suction supernatant down to ~ 5 mL remains. Repeat wash by twice more in 50 mL of S. basal.
- Concentrate worms to ~1000 worms/mL in 7 mL round-bottom glass tube. Add rubber stopper and vacuum seal tube. Add 100 μL of 20% phosphoric acid using a 1 mL syringe with a 25 gauge needle through the rubber stopper to release dissolved worm CO2.
- Remove 2 mL atmosphere and inject into 12 mL blue-top auto-sampler tubes pre-filled with helium for dissolved CO2 measurement.
- Analyze “dissolved CO2 samples” by Gas-Ratio Mass Spectrometer (ThermoQuest Finnigan Delta Plus).
Protocol D. Processing neutralized samples from Protocols A and B for amino acid and organic acid analysis by gas chromatography/mass spectrometry.
- Bead Preparation:
- Add 1 N HCl to both Ag 1 and Ag 50 beads separately in 1L beakers.
- Stir each flask for 30 minutes with magnetic stirrer.
- Wash beads with deionized water 10 times until pH of washings are equal to pH of the water.
- Column Preparation:
- Insert a cotton plug just above the narrowest part of Pasteur pipette tip.
- Add charged beads to each of two columns per sample, filling each to approximately one-third of the column height. Use AG1 beads for organic acid extraction. Use AG50 beads for amino acid extraction.
- Sample Processing:
- Add 500 μL of sample to the column with charged AG1 beads. Nothing is added to the sample (see PROTOCOL A, step #19B) before applying to AG1 column to extract organic acids, if sample is already at neutral pH.
- Add 1 mL of 0.1 N HCL to the remaining sample before applying it to the AG50 column to extract amino acids.
- Allow samples to run completely through columns.
- Wash column with water 10 times until washings are neutral (7-8 pH range).
- Elute columns to fresh, labeled glass 4 mL sample vials:
- For Organic Acid extraction, add 3mL of 3 N HCl to AG1 column. This permits analysis of isotope enrichment in total number of labeled carbons among 3-carbon species (lactate), 4-carbon species (aspartate, succinate, malate), 5-carbon species (glutamate), and 6-carbon species (citrate).
- For Amino acid extraction: add 3mL of 4 N NH4OH to AG50 column. This permits analysis of isotope enrichment in total number of labeled carbons among 3-carbon species (alanine) and 5-carbon species (glutamine).
- Place the sample vials in Reacti-Vap III Evaporator overnight until dry.
E. Sample Derivitization and Machine Settings for Mass Spectrometry
Add 50 μL of acetonitrile and 50 μL of n-methyl-n-t-butyldimethylsilyl trifluoroacetamide (MTBSTFA) to each sample vial. Cover, mix and incubate for 30 minutes at 60°C. Inject 1-2 μl per sample into Gas Chromatography/Mass Spectrometer (GC/MS).
F. Analysis of Results
- Quantification of amino acid peaks. Quantitation of each amino acid by HPLC analysis is calculated using the area of each peak relative to that of the internal standard.
- Calculating isotopic enrichment. Stable isotopic enrichment is calculated in Excel (Microsoft) for each species according to the following formula: Atom Percent Excess, corrected (APE) = (Rsa-Rst)*100/[(Rsa-Rst)+100], where Rsa – Ratio of the sample and Rst – Ratio of the standard.
- Assessing statistical differences between sample enrichment. Student’s t-test is used to assess significance of relative amino acid and isotopic label differences between samples.
REPRESENTATIVE RESULTS:
Stable isotope is well-incorporated into young adult worms, as evidenced by labeled carbon measured in atmospheric CO2 and dissolved worm CO 2. In worms fed either living or UV-irradiated killed OP50 E. coli, the ratio of 13CO2 à 12CO2 was greater in the dissolved worm CO2 fraction relative to the released atmospheric CO2 (Figure 1). Feeding worms live or killed OP50 E. coli did not significantly alter the measured 13CO2 à 12CO2 ratio in washed worm extract (see PROTOCOL C1).
Prolonged exposure to stable isotope from the early larval period increased isotopic enrichment in intermediary metabolites. Corrected Atoms Percent Excess of labeled carbon determined in intermediary metabolites of young adult wild-type worms was greater across all glutamate species when worms were fed universally-labeled 13C-glucose and live bacteria throughout development relative to similarly treated animals fed labeled bacteria for 48 hours only after reaching the egg-laying adult stage (Figure 2).
Effect of clearing times on isotopic enrichment. Regardless of exposure timecourse, all animals grown on plates were ‘cleared’ of isotopic label prior to downstream preparation for GC/MS analyses. Comparison of clearing protocols was performed as shown in Figure 2, including feeding worms for 2 hours on NGM agar plates spread with live fresh bacteria without isotope or feeding on NGM agar plates without bacteria for 2 hours, and feeding on NGM agar plates without bacteria for either 2 or 6 hours. Regardless of isotopic exposure course, no significant difference in isotopic enrichment in intermediary metabolites from young adult whole worm extract was observed when animals were cleared on plates without bacteria for either 2 or 6 hours. By contrast, less isotopic enrichment was observed in intermediary metabolites when animals were subsequently ‘cleared’ of excess isotope by feeding on unlabeled bacteria for two hours. However, increased enrichment in +3, +4, and +5 species was evident when worms were exposed to isotope from the early larval period. Therefore, the optimal clearing protocol was determined to be clearing worms following their incubation on NGM plates spread with stable isotope and bacteria by subsequent incubation for 2 hours on unspread NGM plates. Of note, worms grown in liquid culture were washed clear of isotopic label and bacteria in three volumes of S. basal (see Protocol B); GC/MS analysis of washings showed no significant isotopic enrichment by the third wash.
Worm grinding yielded maximal enrichment in intermediary metabolites. To optimize percent enrichment in intermediary metabolites following similar treatment conditions, comparison was made of worm samples disrupted by sonication alone or sonication plus grinding. Figure 3 shows that worms disrupted following isotope exposure by sonication plus grinding had greater isotopic enrichment than samples disrupted only by sonication. These data suggest that more sub-organismal fractions were disrupted by grinding than by sonication. Subsequent studies revealed grinding alone without sonication was sufficient to achieve maximal isotopic enrichment (data not shown).
Whole worm isotopic exposure permits analysis of intermediary metabolic pathway flux. Feeding living worms with stable isotopic precursor either during development (PROTOCOL A, Figure 4A) or beginning in adult stage animals (PROTOCOL B, Figure 4B) permits sensitive analysis of isotopic enrichment among metabolites indicative of flux through glycolysis (lactate, alanine), pyruvate metabolism (alanine), and the tricarboxylic acid cycle (citrate, malate, succinate, aspartate, glutamate, and glutamine). Universally-labeled 13C-glucose provides robust labeling of all carbon species, whereas utilization of 1,6-13C2-glucose permits robust labeling only in +1 species of each metabolite. This technique can sensitively discern differences from wild-type worm intermediary metabolic flux among mitochondrial respiratory chain mutant strains, such as the complex III mitochondrial respiratory chain subunit mutant isp-1(qm150). Figure 5 illustrates the magnitude of differences observed in absolute label between isp-1(qm150) and N2 worms each exposed for 24 hours to 1,6-13C2-glucose in liquid culture with K12 E. coli bacteria from the first day of egg-laying young adult stage (Protocol B). Additional stable isotopic studies in a range of mitochondrial mutant worms are underway.
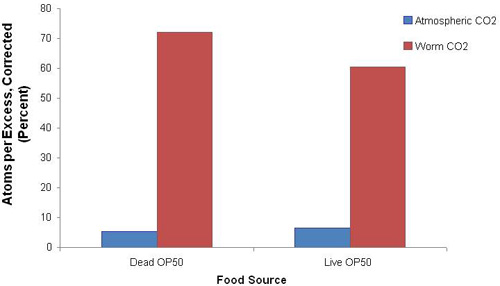
Figure 1. Universally-labeled 13C-glucose stable isotope was well-incorporated into young adult worms. 13CO2:12CO2 ratio (atoms percent excess (APE), corrected) in wild-type (N2) worms following two hours feeding universally-labeled 13C-glucose and either UV-killed or live OP50 bacteria on plates (PROTOCOL C1). 13C incorporation into worm metabolites was robust after two hours of isotope exposure. Blue and red bars indicate relative isotopic enrichment in gas phase (“atmospheric CO2“) and liquid phase (“worm CO2“), respectively.
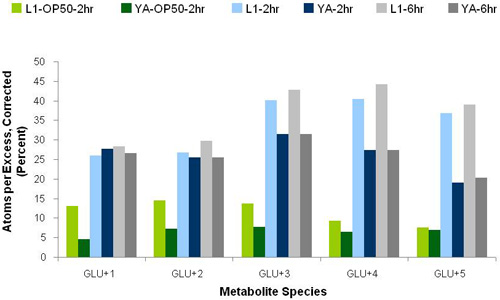
Figure 2. Prolonged isotope exposure from the early larval period and subsequently ‘clearing’ worms on NGM agar plates without bacteria increased isotopic enrichment in free glutamate of young adult worms. Isotopic enrichment in glutamate species is shown for worms fed on NGM agar plates with universally-labeled-13C-glucose and OP50 bacteria either throughout development from the L1 larval stage through the 959 cell young adult stage (“L1”, see PROTOCOL A), or for forty-eight hours beginning when worms reached the first day of egg-laying young adult stage (“YA”). X-axis indicates the total number of labeled carbon atoms in each glutamate species. Y-axis indicates the percent enrichment (corrected atoms per excess (APE)). Green bars indicate worms cleared following isotope exposure by feeding OP50 E. coli without isotope on NGM plates for two hours prior to PCA extraction. Blue and gray bars indicate worms cleared following isotope exposure on NGM plates without bacteria or isotope for two or six hours, respectively, prior to PCA extraction.
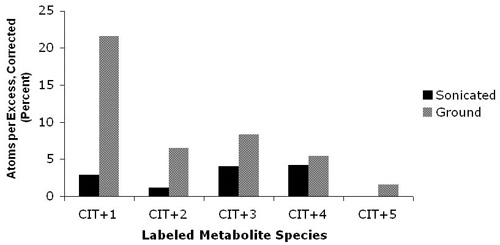
Figure 3. Disrupting worms by grinding yields maximal isotopic enrichment in intermediary worm metabolites. Percent of carbon enrichment measured in +1 citrate species in wild-type worms treated for 24 hours in liquid culture with 1,6-13C2-glucose without bacteria (PROTOCOL B). Black and gray bars indicate worms that were disrupted by sonication only or by sonication and grinding, respectively. Isotopic enrichment following exposure to 1,6-13C2-glucose is best assessed in metabolites labeled on one carbon. In contrast, label is enriched in all species of each metabolite when universally-labeled-13C-glucose is utilized.
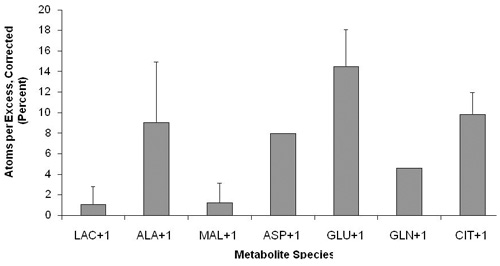

Figure 4. Representative results illustrate the extent of isotopic enrichment in worm intermediary metabolites indicative of flux through glycolysis, pyruvate metabolism, and the tricarboxylic acid cycle. Corrected atoms percent excess (APE) was determined in young adult wild-type (N2 Bristol) worms following 1,6-13C2-glucose exposure either (A) during development from L1 stage on plates (PROTOCOL A), or (B) beginning on the first day of egg-laying as young adults for 24 hours in liquid culture (PROTOCOL B). Error bars indicate standard deviation where reliable isotopic data from three biological replicates was available.
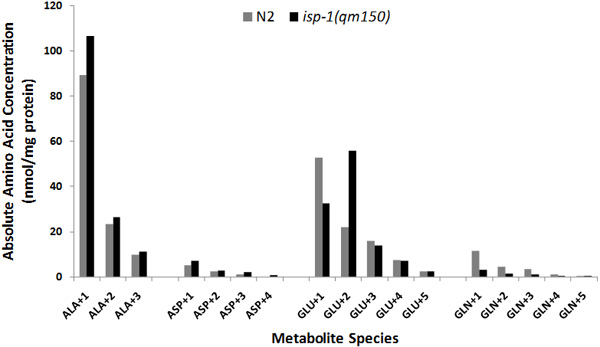
Figure 5. Absolute amino acid quantitation of amino acid metabolite species in wild-type and mitochondrial mutant worm strains. Absolute label in each species of four amino acids was calculated by multiplying HPLC-determined free amino acid concentration (nmol/mg worm protein) by corrected atoms percent excess (APE) for each species. Gray and black bars indicate wild-type (N2 Bristol) and mitochondrial complex III subunit mutant strains (isp-1(qm150)), respectively. Bars indicate average of three biological replicate experiments per strain.
