1. Mouse infection with γHV68
- Six-to-eight-week old, gender-matched littermate mice (8 to 12 mice/group) are used for viral infection. Allow mice to acclimate over four full days (96 hours) after shipment.
- Protocol steps using virus should be carried out in a cabinet of biosafety level 2 (BSL2) using standard BSL2 precautions.
- Prepare viral suspension (40 to 1 x 105 plaque-forming units [PFU]) of γHV68 in 30 μl of sterile PBS per mouse just before the experiment. Keep viral suspension on ice.
- Prepare ketamine/xylazine solution (1.5 mg ketamine and 0.15 mg xylazine/20 g body weight, 100 μl/mouse) for mouse sedation. Ensure that the mice have been sedated by performing a toe pinch.
- Sedate mice with 1.5 mg ketamine and 0.15 mg xylazine per 20 mg body weight (100 μl/mouse) by intra-peritoneal injection.
- Deliver viral suspension (30 μl/mouse) intranasally, in a drop-wise fashion, to one nostril of the sedated mice.
- Lay mice on one side for 5 – 10 min to facilitate the airway delivery of the virus into the lung.
- Place mice back in cage and monitor mice until they have fully recovered from sedation.
- At various days post-infection, sacrifice mice by CO2 asphyxiation and harvest the lungs after assuring death/loss of deep consciousness. Place lungs into sterile 1.5 ml screw-capped tubes containing 500 μl of 1.0 mm Zirconia/Silica beads. Keep tubes on ice. Store samples at -80°C if not proceeding to next step on the same day. The spleen or liver tissue could be collected at this time for the analysis of viral latency depending on time frame of experiment.
- Add 1 ml of cold serum-free DMEM into the tube and homogenize the lungs by bead-beating 30 seconds. Chill tubes on ice for at least 1 min. Repeat this process once.
- Centrifuge lung lysates at 16,000 rcf at 4°C for 1 min and use supernatant to determine viral titer by a plaque assay using a NIH3T3 or BHK21 monolayer (see Section 5 for details).
2. Determine γHV68 multi-step growth kinetics in mouse embryonic fibroblasts
- Grow wild-type mouse embryonic fibroblasts (MEFs) and those deficient in a host gene to sub-confluent (approximately 80%) density before plating cells.
- Split MEFs into 24-well plate at 10,000 cells/well for a low multipilicity-of-infection (MOI) on the day before infection. Experiments are normally carried out in triplicates and at a MOI between 0.001 – 0.05 is usually used.
- Prepare γHV68 suspension containing desired amount of virus (0.5 ml per well).
- Remove medium and cover MEFs with 0.5 ml of γHV68 suspension per well.
- Incubate the plate in tissue culture incubator, rock every 30 min, and allow incubation to proceed for 2 h.
- Remove viral suspension, and cover cells with 0.5 ml fresh complete DMEM medium containing 8% fetal bovine serum.
- At various days post-infection, harvest the medium and cells into sterile 1.5 ml centrifuge tubes. Immediately freeze tubes at -80°C.
- Release γHV68 from MEFs by freezing at -80°C , thawing in 37°C water bath and vortexing. Three cycles of freezing and thawing is usually applied to the samples.
- Determine viral titer by a plaque assay using a NIH3T3 or BHK21 monolayer (see Section 5 for details).
- Read viral titer and plot γHV68 multi-step growth curve on MEFs.
3. Molecular dissection of γHV68 lytic replication in mouse embryonic fibroblasts
- Perform viral infection as described in steps 2.1 to 2.6 of section 2.
- At various days post-infection, discard the supernatant. Rinse cells with cold PBS and trypsinize cells. Pellet cells by centrifuge at 1,000 rcf at room temperature for 1 min. Discard the supernatant and store cells at -80°C.
- Extract total DNA (host and viral genome) and total RNA according to previously published methods5,6.
- Perform real-time PCR using total DNA and primers specific for viral lytic transcripts, such as RTA (ORF50), ORF57 and ORF60. Determine the relative quantity of intracellular γHV68 genome in reference to a host housekeeping gene (e.g., β-actin).
- To remove genomic DNA contamination, treatment with RNase-free DNase is critical for cDNA preparation with total RNA and oligo(dT)11-19 primer. Refer to reference 5 for more details on the RNA extraction including treatment with RNase-free DNase and RT-PCR. Perform real-time PCR using cDNA and primers as above to determine the relative quantity of viral transcripts in reference to that of host housekeeping gene.
4. Generating recombinant γHV68 using bacterial artificial chromosome
The method described here is used to introduce mutations into a γHV68 gene that is involved in host-virus interaction.
- Prepare a DNA fragment (about 1.5 kb) of wild-type sequence or sequence carrying desired mutations in the central region by PCR.
- Prepare bacterial artificial chromosome (BAC) that contains a transposon insertion7 around the mutation site, which specifically inactivates the gene gene of interest, by midi-scale purification (OriGene). Store BAC DNA at 4°C (avoid freezing/thawing of BAC DNA).
- Transfect BAC DNA and PCR product containing desired mutations of the gene of interest into cells (e.g., NIH3T3 or BHK21), which are highly permissive to γHV68 lytic replication, with Lipofectamine 2000 (Invitrogen).
- Keep splitting cells until cytopathic effect (CPE) shows up in the monolayer. Collect virus-containing supernatant and, if necessary, amplify virus in NIH3T3 or BHK21 cells.
- Infect NIH3T3 cells with recombinant virus by centrifugation at 1,800 rpm, 30°C for 30 min.
- Harvest γHV68-infected NIH3T3 cells and prepare circularized viral genome using Hirt’s protocol8,9.
- Transform Electro-MAX DH10B competent cells (Invitrogen) by electroporation and screen for colonies that are resistance to chloramphenicol (Cm), but sensitive to kanamycin (Kan) (Cm-resistant gene is in BAC backbone, while Kan-resistant gene is in transposon insertion).
- Grow CmRKanS colonies in medium containing chlorophenicol and prepare BAC DNA with mini- or midi-scale purification (OriGene).
- Verify the desired mutation in the target gene by PCR, using primers specific to flanking regions of mutation site, and sequencing.
- Confirm no chromosome rearrangement in BAC by digestion with selected restriction enzymes and pulse-field gel electrophoresis.
- Transfect recombinant BAC into NIH 3T3 or BHK21 cells with Lipofectamine 2000 (Invitrogen) to prepare recombinant γHV68.
- Keep passaging cells until CPE shows up in the monolayer. Collect virus-containing supernatant and amplify recombinant γHV68 in NIH3T3 or BHK21 cells.
- Determine titer of the recombinant virus and characterize viral growth properties ex vivo and in vivo as described in sections 1 and 2.
5. Determine viral titer by a plaque assay
- Grow NIH3T3 cells to sub-confluent (approximately 80%) density before plating cells.
- Split NIH3T3 into 24-well plate at 20,000 cells/well on the day before infection.
- Prepare 10-fold serially-diluted virus supernatants with DMEM medium containing 8% newborn calf serum (NCS).
- Remove medium and cover NIH3T3 cells with 0.5 ml of γHV68 suspension.
- Incubate the plate in tissue culture incubator, rock every 30 min, and allow incubation to proceed for 2 h.
- Remove viral suspension and cover cells with 0.5 ml DMEM medium containing 2% NCS and 0.75% methylcellulose (Sigma).
- Incubate the plate in tissue culture incubator. Count plaques at day 6 post-infection. Staining of the monolayer, e.g. with crystal violet, may facilitate plaque counting.
- Calculate the viral titer in the undiluted tissue lysates or cell lysates using the following formula: Titer (PFU/ml) = D x N x 1000 μl/ml ÷ V μl. N, the mean of plaque number at an appropriate dilution; D, dilution fold (such as 5, 10, 100…..) V (μl), volume of serially-diluted supernatant added per well.
6. Representative Results:
Three representative figures are shown here, including γHV68 lytic replication in the lung of wild-type and Mavs-/- mouse10, γHV68 lytic replication phenotypes in mouse embryonic fibroblasts (MEF), and recombinant γHV68 carrying mutations within the phosphorylation sites that are modulated by the MAVS-dependent IKKβ. These three corroborating experiments constitute a scheme to define the roles of innate immune components in γHV68 lytic replication in vivo and ex vivo.
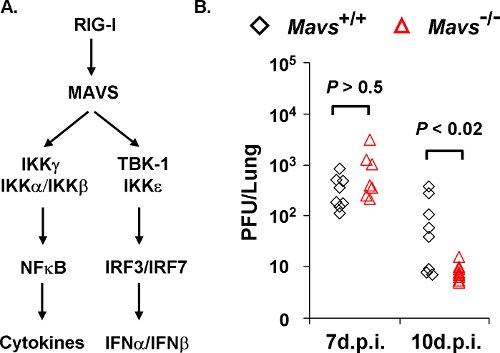
Figure 1. γHV68 loads in the lungs of Mavs+/+ and Mavs-/- mice. A) Two main innate signaling pathways downstream of MAVS. The MAVS adaptor molecule relays signaling from cytosolic RIG-I-like receptors to activate NFκB and interferon regulatory factors (IRFs) that, in turn, up-regulate the gene expression of proinflammatory cytokines and interferons. B) Mavs+/+ and Mavs-/- mice were infected with 40 PFU γHV68 intranasally and viral loads in the lung at indicated time points were determined by a plaque assay using NIH3T3 monolayer. Each symbol represents a mouse.
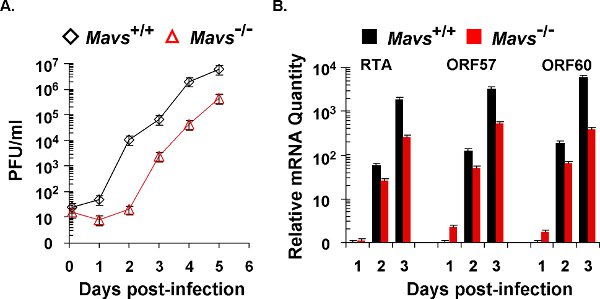
Figure 2. γHV68 lytic replication kinetics in mouse embryonic fibroblasts (MEFs). The lytic replication of γHV68 on Mavs+/+ and Mavs-/- MEFs was assessed by multi-step growth curves (A) and quantitative real-time PCR (B). For both experiments, equal number of MEFs and amount of γHV68 were used for viral infection at a multiplicity-of-infection (MOI) of 0.01. (A) Cells and supernatants were harvested at indicated time points and subject to a plaque assay to determine viral titers. (B) Total RNA was extracted from γHV68-infected MEFs and analyzed by quantitative real-time PCR with primers specific for selected lytic transcripts (RTA, ORF57, and ORF60).
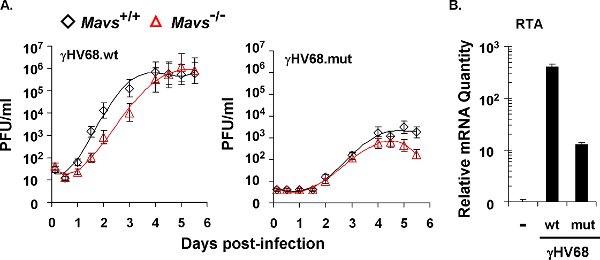
Figure 3. The lytic replication kinetics of recombinant γHV68 carrying mutations within the RTA transactivation domain that abolish phosphorylation by IKKγ. (A) Multi-step growth curves of recombinant wild-type virus (γHV68.wt) and mutant virus (γHV68.mut) in Mavs+/+ and Mavs-/- MEFs cells (MOI=0.01). MEFs were infected with γHV68 at an MOI of 0.01. Cells and supernatants were harvested at indicated time points and viral titers were determined by a plaque assay using NIH 3T3 monolayer. (B) γHV68 RTA mRNA level in γHV68-infected NIH3T3 cells (MOI=0.01). At 30 h post-infection, total RNA was extracted from γHV68-infected NIH 3T3 cells and analyzed by quantitative real-time PCR.
