Ethics Statement
The mice used in this work were cared for according to Canadian Council on Animal Care (CCAC) guidelines. Ethical approval for experiments conducted was obtained from the University of Ottawa Animal Care Committee under protocol number OGH-119.
1. Dissection – neonatal mouse cortex for OPC extraction
- Sacrifice P0-P2 mouse according to institutional guidelines.
- Dissect the brain and place in a Petri dish containing ice-cold MEM (antibiotic-free).
- Transfer the dish to a dissection microscope.
- Using a scalpel with the brain dorsal side up, make a shallow incision sagittally along the most medial edge of each cortex (Fig 1a). This incision should only pass through the meningeal layer in order to facilitate its removal.
- Use fine tipped forceps to peel off the meninges in a lateral fashion. If done carefully, this layer can be removed in one piece. During this step, remove the olfactory bulbs.
- With the brain ventral side-up, make a deep sagittal incision where the cortex meets the ventral area of the diencephalon (Fig 1b).
- With the brain dorsal side-up, separate the cortices from the midbrain by prying the tissue in a medial to lateral fashion (Fig 1c, c’). Remove any residual meninges at this step.
- Dice each cortex into approximately 4 pieces and gently transfer to a 15 mL conical tube containing 350 μL of MEM per mouse brain. Keep the tube on ice until all mice have been processed.
- Repeat steps 1.1-1.8 for remaining mice.
2. Dissociation of neonatal cortices and maintenance of mixed glial cultures
Note: The introduction of bubbles into the cell suspension should be avoided during all of the following steps.
- Add the 15 mL conical tube containing the freshly dissected brains to a 37°C water bath for 3 min.
- Transfer brains to a sterile tissue culture hood.
- Gently pass diced cortices through a P1000 pipette tip to generate smaller fragments. Stop pipetting once there are no brain pieces large enough to disrupt a smooth flow of suspension through the pipette tip.
- Add 75 μL of OPC papain solution per brain into the conical tube. The OPC papain solution must be pre-warmed at 37°C for 20 min prior to use.
- Incubate in a 37°C water bath for 20 min. Approximately every 2 minutes, gently invert the tube to prevent tissue aggregation. During this time, add 5 mL of mixed glial culture media to each poly-L-lysine (PLL) coated (1 mg/mL) T25 flask (one flask per mouse brain), and place in a 37°C tissue culture incubator at 8.5% CO2.
- After 20 min, return the tissue suspension to the sterile hood and add 2 mL of mixed glial culture media per brain to the tube. Let sit for 10 min at room temperature to allow inactivation of the OPC papain solution.
- Aliquot the tissue suspension into 5 mL plastic tubes. The number of tubes should match the number of brains dissected, resulting in approximately 2.5 mL per tube.
- Using a sterile flame-polished glass Pasteur pipette, gently triturate the tissue in each tube. Triturate slowly at first, and gradually increase speed as pieces dissociate. Triturate approximately 10-15 times, however this number may vary based on the efficacy of the digestion.
Note: Under-trituration will result in poor dissociation of the tissue, whereas over-trituration will negatively impact on cell viability. It is important to not introduce bubbles into the solution, as this will severely impact cell viability.
- Once there are no visible tissue clumps remaining in the suspension, transfer to a 50 mL conical tube containing 4 mL of mixed glial culture media per brain (i.e., 4 brains = 16 mL mixed glial culture media).
- Gently invert the 50 mL conical tube and repeat for the remaining 5 mL tubes.
- Aliquot the pooled cell suspension into 15 mL conical tubes (approximately 6.5 mL per 15 mL tube). The number of 15 mL tubes should match the number of brains dissected.
- Centrifuge the tubes at 1200 rpm (~300 g) for 5 min.
- Carefully aspirate the supernatant and add 1 mL of warm mixed glial culture media to each 15 mL conical tube.
- Slowly resuspend the pellet with a P1000 pipette tip, being careful not to introduce bubbles. Add the cell suspension from each tube to a pre-equilibrated PLL-coated T25 flask, rendering the total volume of the culture media to 6 mL.
- Place the flasks in a tissue culture incubator for 3-4 hours to allow the cells to attach to the PLL substrate. Perform a full media change by pipetting out the media, and adding 6 mL of fresh mixed glial culture media to the flasks. This step removes much of the debris caused by the trituration, and promotes culture viability. If OL/DRGN co-cultures are desired, refer to Section 3 of this protocol.
- After 3 days of culture, perform a 2/3 media change by removing 4 mL of media, and replacing with 4 mL of fresh mixed glial culture media. At this point, an astrocyte monolayer should be forming on the base of the flasks.
- On Day 6, perform another 2/3 media change and supplement the flasks with a final concentration of 5 μg/mL insulin. At this point, an astrocyte monolayer should be clearly visible, on top of which OPCs will be proliferating.
3. DRGN isolation
Note: To produce OL/DRGNs co-cultures, DRGNs should be established the day after mixed glial culture generation. Both culture types are grown independently, and combined after 9-10 days.
- Sacrifice P5-P10 mouse according to institutional guidelines.
- Extract the spine, and transfer to a clean Petri dish.
- Trim away as much muscle and bone from the spine as possible (Fig 1d, d’), as this will ease the dissection of the dorsal root ganglia (DRGs).
- Transfer the trimmed spine to a new Petri dish ventral side-up. Using dissection scissors and starting caudally, cut medially through the spinal column in a longitudinal fashion.
- Using two pairs of forceps, gently pry open the spinal column to expose the spinal cord.
- DRGs can be found beneath and lateral to the spinal cord. Using fine tipped forceps, gently remove the DRGs while avoiding damage to the ganglia (Fig 1e).
- Transfer the removed DRGs to ice cold Hank’s buffered salt solution (HBSS, antibiotic-free) in a new Petri dish. The dissector should aim to extract 40 DRGs per mouse (Fig 1f).
- Once the DRGs have been extracted, trim the DRGs of any excessively long roots (Fig 1g, g’) to minimize introduction of contaminating cells into the culture (glial cells, fibroblasts).
- Transfer the DRGs to a 1.5 mL centrifuge tube containing 500 μL of ice cold HBSS.
- Centrifuge at 1200 rpm (~300 g) for 5 min at 4°C to pellet the DRGs.
- Transfer the centrifuge tubes to a sterile tissue culture hood and remove the HBSS from the tubes.
- Add 500 μL of pre-warmed (20 min at 37°C) DRG papain solution, and incubate the tubes in a 37°C water bath for 10 min. Invert the tubes every 2 min to prevent tissue aggregation.
- Repeat step 3.10.
- Remove the DRG papain solution and add 500 μL of pre-warmed (20 min at 37°C) Collagenase A solution. Incubate in a 37°C water bath for 10 min, inverting every 2 min.
- Repeat step 3.10.
- Remove supernatant and add 1 mL of DRGN media. Invert tube several times.
- Repeat step 3.10.
- Repeat step 3.16.
- Coat a sterile flame-polished glass Pasteur pipette with bovine serum albumin (BSA) by pipetting a solution of 0.25% BSA in HBSS several times. The coating with BSA solution will prevent the DRGs from adhering to the walls of the glass pipette.
- Triturate the DRGs with the BSA-coated pipette gently at first, and with increasing intensity once clumps begin dissociating. Triturate approximately 10-15 times, however this number is dependant on the degree of digestion, and the number of DRGs per tube.
- Once dissociation is achieved, pass the suspension through a 50 μm filter into a sterile Petri dish containing 7 mL of DRGN media. Filtration will eliminate much of the debris from the cell suspension, although this step is not critical.
- Incubate the Petri dish at 8.5% CO2 for approximately 1.25 hours.
- Coat several 12 mm coverslips with LN2 (10 μg/mL in PBS) in a 24-well dish during this incubation time.
- Once the incubation is finished, observe the Petri dish under bright field. DRGNs are identified as large bodied, phase dark cells. Swirl the Petri dish gently to lift any adhered DRGNs.
Note: Many contaminating cells will have strongly adhered to the Petri dish, thereby enriching your cell suspension for DRGNs.
- Transfer the cell suspension to a 15 mL conical tube. Gently rinse the dish with 4 mL of DRGN media to collect any residual DRGNs. Transfer the additional 4 mL to the conical tube.
- Centrifuge for 5 min at 1200 rpm (~300 g).
- Aspirate supernatant and resuspend the pellet in 500 μL of fresh DRGN media.
- Calculate the number of yielded DRGNs using a hemocytometer. Be sure to only count the DRGNs, and not other cell types. DRGNs can be identified by their large spherical cell bodies.
- Seed 30,000–50,000 DRGNs to each LN2-coated coverslip in 1 mL of DRGN media, and place in a 37°C tissue culture incubator at 8.5% CO2 overnight.
- The next morning, perform a full media change by replacing the DRGN media with OL media (minus CNTF) with a final concentration of 1% Pen/Strep and 10 μM FuDR.
- On Days 3 and 5, perform a 3/4 media change with the same media as in Step 3.30.
- On Day 7, perform a full media change with OL media (minus CNTF, Pen/Strep, FuDR).
- On Day 9, the DRGNs should have formed an extensive neurite bed, and are now ready to be co-cultured with OPCs.
4. Purification of OPCs from mixed glial cultures for establishment of OL-enriched cultures or OL/DRGN co-cultures
- On Day 9 of the mixed glial culture, transfer the flasks to an orbital shaker in a 5% CO2 tissue culture incubator. Place the flasks on top of empty T25 flasks to prevent any heat generated from the orbital shaker from adversely affecting the mixed glial cultures. Allow the cultures to equilibrate to this new incubator for 1 hour.
- Once the flasks have equilibrated, shake the flasks at 50 rpm for 45 min. The purpose of this shake is to remove any loosely adherent contaminating cells from the monolayer.
- Move cells to a tissue culture hood and remove all the media from the flasks. Replace with 4 mL of fresh mixed glial culture media supplemented with 5 μg/mL insulin.
- Place the flasks back onto the shaker, and allow to equilibrate for approximately 3 hours.
- Once the flasks are equilibrated, fasten them securely to the orbital shaker, and shake the flasks for approximately 16 hours at 220 rpm (overnight).
- The next morning, if OLs are to be grown in the absence of DRGNs (i.e., OL-enriched culture), coat several sterile 12 mm coverslips with LN2 (10 μg/mL in PBS) for 1 hour. Transfer the coverslips to 24-well dishes, wash with PBS followed by an OL media wash. Add 1 mL of OL media to each well and equilibrate at 8.5% CO2.
- Equilibrate 10 cm tissue culture dishes at 5% CO2 for 30 min. One dish will be required for every 2 flasks. These will be used for the differential adhesion-enrichment of the suspended OPCs.
- Once the 30 min equilibration period has passed, transfer the media from the shaken flasks to the dishes. Each dish should receive media from 2 flasks, equaling approximately 8 mL of cell suspension per 10 cm dish.
- Incubate the dishes at 5% CO2 for 30 min, while providing a gentle nudge at the 15 min mark. This nudge will help prevent OPCs from adhering to the 10 cm dish.
- Once the incubation is complete, examine the dishes under bright field. OPCs are identified as small cell clumps, typically of 3-5 cells but sometimes form large aggregates resembling neurospheres. Many non-OL lineage cells should be firmly adhered to the base of the plate. Gently swirl the plates to detach any loosely adhered OPCs, and transfer the cell suspension from each plate into a 15 mL conical tube.
- Centrifuge at 1200 rpm (~300 g) for 5 min.
- Resuspend the pellet in 1 mL of OL media with a P1000 pipette tip, followed by resuspension with a P200 pipette tip.
- Count the cells using a hemocytometer.
- For enriched-OL cultures, seed 25,000 – 50,000 OPCs to each 12 mm LN2-coated coverslip in a final volume of 1 mL OL media.
- For OL/DRGN co-cultures, perform a full OL media (minus CNTF) change on the DRGNs from section [3], and gently add 50,000 cells from the OPC-enriched cell suspension. Take care to not disrupt the DRGN neurite bed during the addition of OPCs.
- Place cultures in a 37°C incubator at 8.5% CO2, and avoid removing until fixation. Murine OPCs are sensitive to changes in pH, and removal from the incubator will alter the pH of the OL media. Also of note, addition of dH2O to the empty wells surrounding the cell cultures will prevent evaporation of the culture media, thus minimizing fluctuations in the concentrations solutes within the OL media. This will provide a more consistent environment for the OPCs.
5. Processing of cultures for immunofluorescence microscopy
- Fix cultures with 100% methanol at -20°C for 10 min, or 3% paraformaldehyde at room temperature for 15 min.
- Permeabilize coverslips with 0.1% Triton-X-100 for 10 min, wash with phosphate-buffered saline (PBS) and block for 1 hour in 10% goat serum.
- Incubate coverslips with primary antibodies diluted in blocking solution overnight at 4°C.
- Wash coverslips 3 times with PBS, and incubate with Alexa-fluor conjugated secondary antibodies (Invitrogen) diluted in blocking solution for 45 min.
- Counterstain with 4′,6-diamidino-2-phenylindole (DAPI) and wash coverslips several times with PBS.
- Mount coverslips in DAKO fluorescent mounting medium.
- Analyze slides via immunofluorescence microscopy. In this protocol, slides were analyzed with either a Zeiss Axiovert 200M inverted fluorescence microscope or a Zeiss LSM 510 META laser scanning confocal microscope.
6. Whole cell protein extraction from OL-enriched cultures
- Remove 24-well cultures from incubator and cool on ice for 3 min.
- Carefully remove media, and add 10-20 μL of lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton-X-100, with 0.1% pepstatin, aprotinin, PMSF, leupeptin, sodium orthovanadate) to each well (A minimum of 8 wells per sample is suggested).
- Scrape wells using a wide-bore P1000 pipette tip, and transfer the lysate to a 1.5 mL centrifuge tube.
- Pass the lysate through a 30½-gauge syringe approximately 15 times, and chill on ice for 30 min.
- Centrifuge tubes at 14,000 rpm (~20,000 g) for 15 min at 4°C.
- Transfer supernatant to new centrifuge tubes, and store at -80°C.
7. SDS-PAGE analysis on enriched-OL culture protein
- Resolve 30 μg of protein per sample in reducing buffer by SDS-PAGE on standard 12% poly-acrylamide gels.
- Semi-dry transfer gels onto PVDF membranes.
- Block membranes for 1 hour in 5% skim milk powder in TBST (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.1% Tween-20).
- Incubate membranes with primary antibodies diluted in blocking solution for 1 hour.
- Wash membranes 3 times with TBST for 10 min.
- Incubate the membranes with HRP-conjugated secondary antibodies for 45 min in blocking solution.
- Wash membranes several times with TBST, and incubate with Amersham ECL Plus western blotting detection reagent (GE Healthcare) for 5 min.
- Detect protein bands with standard scientific imaging film.
8. Representative Results:
In this protocol, OPCs are expanded on an astrocyte monolayer within a mixed glial culture. This mixed glial culture is derived from P0-P2 neonatal mouse cortex. At day 1 in vitro (DIV1), the mixed glial culture contains cells with varying morphologies as seen by phase contrast microscopy (Fig 2a). At DIV3, an astrocyte monolayer begins to form on the base of the flask, and at DIV8, OPCs can be clearly observed on the monolayer surface. At DIV9, the proliferating OPCs have reached sufficient density to be purified by overnight high-speed orbital shaking. Once the purification process has been completed, the result is an OPC-enriched cell population. At DIV1-post purification, OPCs have simple morphology, extending few processes (Fig 2b). At DIV3 post purification, cells have extended a complex meshwork of processes, reminiscent of immature OLs. At DIV6 post purification, the purified OLs have flattened and projected leaflet-like membrane structures. This morphological development is typical of the in vitro maturation of OLs.
Immunofluorescence microscopy indicates the purified cells are of OL-lineage (Fig 3a). Seeded OPCs initially express chondroitin sulfate proteoglycan (NG2), and develop into myelin-associated glycoprotein (MAG) positive immature OLs within three days post seeding (Fig 3b). At DIV6, many OLs express myelin basic protein (MBP), and possess typical mature OL morphology. Percent OL-lineage cells were quantified at different time points to determine the purity of the OL-enriched cultures (Fig 3c). At DIV1 post purification, cultures are 50±14% NG2+ve OPCs, with no MAG+ve or MBP+ve OLs. This indicates the purified OL-lineage cells are in the precursor stage at seeding time, with negligible numbers of differentiated OLs. At DIV3, many OLs have differentiated into MAG+ve cells (24±5.9%) while some retain the precursor phenotype, and remain NG2+ (13±8.0%). At DIV3, a small proportion of MAG+ve cells (3.2 1.2%) are also expressing MBP. At DIV6, 20±5.9% of OLs are MAG+ve while 12±7.3% persist as NG2+ve OPCs. In addition, 21±9.3% of cells within the culture are MBP+ve OLs at this time point. SDS-PAGE analysis shows the graded expression of 2’3′-cyclic-nucleotide 3′-phosphodiesterase (CNP) and MBP over the 6 day culture period, further demonstrating the ability of OPCs in culture to terminally differentiate into mature OLs (Fig 3d). Collectively, these data establishes this method as a means of producing an OL-enriched culture system suitable for the study of OL maturation from OPCs.
This protocol also describes methods to establish OL/DRGN co-cultures using murine-only tissue sources. However, in order to produce the co-culture, DRGNs must first be cultured alone to produce an adequate neurite network. These post-natal murine neuron cultures are grown for 9 days in low serum media with 10 μM FuDR supplementation to prevent the proliferation of contaminating fibroblasts and glial cells. Over the course of 9 days in vitro, isolated DRGNs produce a dense neurite bed (Fig 4a). This neurite bed is immunopositive for the neuronal markers neurofilament 200 (NF) and Tuj1 (Fig 4b). At this point, purified OPCs may be added to the neurite beds, and cultured for an additional 6 days to produce myelinating co-cultures.
At DIV6 of OL/DRGN co-culture, many MBP+ve OLs can be observed among the NF+ve DRGN neurites (Fig 5a). Upon closer examination, OLs are evidenced to make contact with numerous DRGN neurites, often ensheathing them with an MBP+ve membrane (Fig 5b,c).
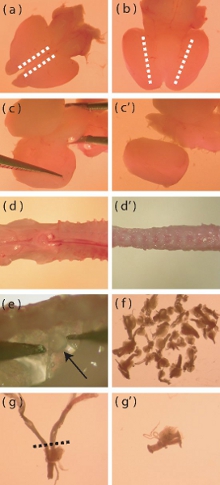
Figure 1. Dissection microscope images of particular aspects of neonatal mouse cortex and DRG isolation. (a) Dorsal view of a freshly extracted neonatal mouse brain. The dotted lines indicate the area where incisions must be made to facilitate the removal of the meningeal layer. (b) Ventral view of brain, dotted lines indicate the area where the cortex meets the ventral diencephalon. Deep incisions must be made along the dotted lines to aid the isolation of the cortices. (c-c’) Visual depiction of how to pry the cortex away from the remainder of the brain. (d) A freshly isolated P5-P10 mouse spine prior to trimming away excess muscle and bone (d’). (e) Location of DRGs within the spinal column. (f) The approximate number of DRGs that should be isolated from one mouse. (g) A DRG with long roots that require trimming prior to enzymatic digestion. The dotted line indicates the region where the roots should be trimmed. (g’) DRG post root-trimming.
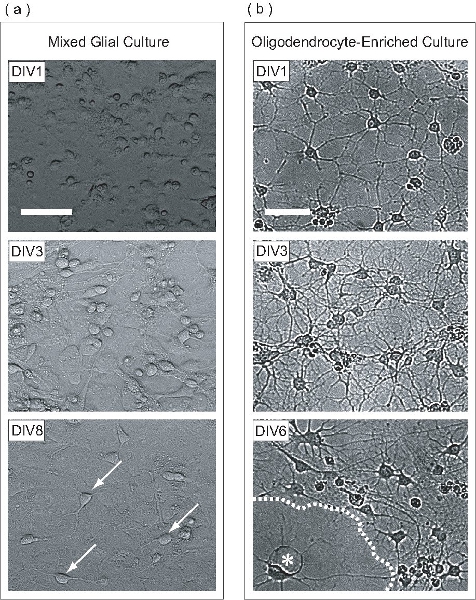
Figure 2. OPCs are expanded within a mixed glial culture, purified, and subsequently differentiated as an OL-enriched culture. (a) Phase contrast images of mixed glial cultures at different stages of development. At DIV1, cells appear round with few flattened cells. Stratification of mixed glial culture begins at DIV3, where astrocytes form a uniform monolayer at the base of the flask, upon which OPCs proliferate. Many OPCs are seen at DIV8 (arrows) adhered to the surface of the astrocyte monolayer. (b) Once purified from the mixed glial culture, DIV1 OPCs have extended only a few processes. At DIV3, cells have extended many processes, reminiscent of intermediate-stage OLs. At DIV6, flattened OLs (asterisk) appear to have produced membranous sheets (dashed line). Scale bars, 50 μm.
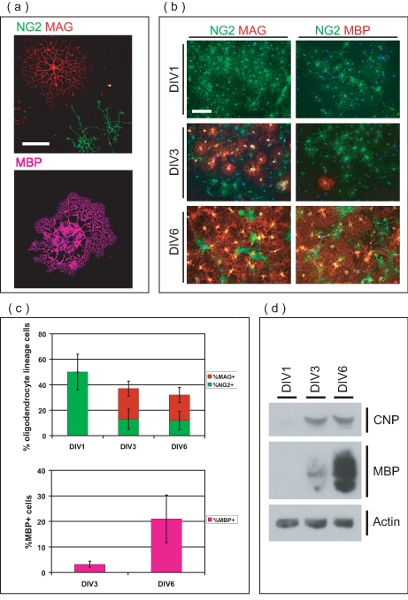
Figure 3. Characterization of OL-enriched culture. (a) Confocal images of isolated OLs at different stages of development. NG2+ve OPCs have simple morphology, whereas MAG+ve OLs possess multiple arborous processes. MBP+ve OLs have extended membranous myelin-like sheets. Scale bar, 50 μm. (b) Purified OL-lineage cells originate as OPCs, and differentiate into MAG+ve, MBP+ve OLs over 6 DIV. At DIV1, all OPCs are NG2+ve, while none are MAG+ve or MBP+ve. At DIV3, MAG+ve and few MBP+ve OLs are now evident. The majority of OLs are MAG and MBP+ve at DIV6, with few remaining NG2+ve OPCs. Scale bar, 100 μm. (c) Mean values ± SD of the percent OL-lineage cells at different stages of development over 6 DIV. At DIV1, all OL-lineage cells are NG2+ve, accounting for 50±14% of total cells within the culture. At DIV3 and DIV6, OL-lineage cells respectively account for 36±6.8% and 32±8.4% of total cells, consisting of varying proportions of NG2+ve, MAG+ve and MBP+ve OLs. (d) SDS-PAGE performed on protein derived from enriched OL-cultures demonstrating the graded expression of OL-markers CNP and MBP over the 6 DIV culture period.
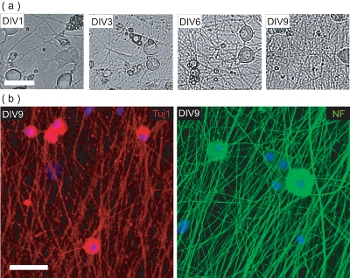
Figure 4. Characterization of the DRGN culture pre-OPC seeding. (a) Phase contrast images of DRGNs over the 9 DIV culture period pre-OPC seeding. DRGNs originate as large-bodied cells with few processes, and produce an increasingly complex neurite network. Scale bar, 100 μm. (b) Confocal images of DRGN cultures fixed at DIV9 (pre-OPC seeding) and stained for neuron-specific markers Tuj1 and NF200. DRGNs have produced a neurite network upon which OPCs may be seeded to produce OL/DRGN myelinating co-cultures. Scale bar, 50 μm.
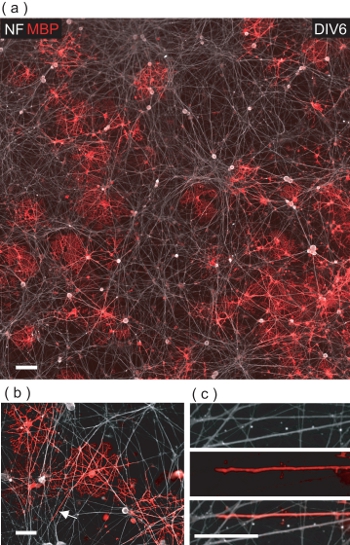
Figure 5. OLs co-cultured with DRGNs result in OL-mediated wrapping of DRGN neurites with MBP+ve membrane. (a) A 4-field confocal image montage of a DIV6 OL/DRGN co-culture. Many MBP+ve OLs can be seen interacting with the underlying DRGN neurite bed. Scale bar, 100 μm. (b) A magnified confocal view of MBP+ve OLs wrapping multiple DRGN neurites. Scale bar, 50 μm. (c) Digital magnification of the region denoted in (b) where a DRGN neurite is being wrapped with OL membrane. Scale bar, 25 μm.
