1. External and internal Solutions
- Prepare slicing solution (low calcium) from 10x stock solution and add MgCl2, CaCl2, and D-glucose daily. The final 1x solution consists of (in mM): 119 NaCl, 2.5 KCl, 3.2 MgCl2, 0.25 CaCl2, 12 D-glucose, 0.2 L-ascorbic acid, 12 HEPES. Set the pH to 7.4 (with NaOH), and adjust osmolarity to 260 mOsm (using H2O and 10x stock solution).
- Weigh out 3% low gelling-temperature agar (Agarose type VII-A, A0701, Sigma; Rieke, 2001) and mix with slicing solution. Heat the agar containing solution in a microwave for 1-2 min, or until it dissolves completely, and incubate the agar in a water-bath (30-33°C) to prevent the rapid solidification (gel transition temperature: 26 ± 2°C).
- Prepare recording solution (normal calcium) from 10x stock solution and add MgCl2, CaCl2, and D-glucose daily. The final 1x solution consists of (in mM): 100 NaCl, 2.5 KCl, 1 MgCl2, 25 NaHCO3, 0.2 L-ascorbic acid, 2.5 CaCl2, 12 D-glucose. After bubbling the solution in 95% O2 / 5% CO2 (carbogen) for 5-10 minutes, set the pH to 7.4 (with NaOH), and adjust osmolarity to 260 mOsm (using H2O and 10x stock solution).
- Aliquots (1 ml each) of internal pipette solutions are prepared in advance and stored in a freezer (-20°C). Two internal pipette solutions are typically used to isolate bipolar cell calcium currents (in mM):
- 40 CsCl, 60 Cs-gluconate, 10 tetraethylammonium (TEA)-Cl, 28 HEPES, 3 Mg-ATP, 1 Na-GTP, and 2 EGTA. Adjust pH to 7.2 (with CsOH) and osmolarity set to 250 mOsm. This high chloride internal solution typically provides lower series resistance recordings (better voltage clamp conditions) and larger inhibitory postsynaptic currents (IPSCs) at a bipolar cell resting membrane potential of -60 mV.
- 95 Cs-gluconate, 10 TEA-Cl, 25 HEPES, 3 Mg-ATP, 0.5 Na-GTP, and 0.5 EGTA. Adjust pH to 7.2 (with CsOH), and set osmolarity to 250 mOsm 22. This low EGTA internal solution typically leads to higher membrane capacitance changes elicited by depolarizing step pulses and thus allows studies of vesicle pool depletion and short-term depression.
2. Patch-clamp pipette electrodes
- Prepare thick-walled (1.5 mm outer diameter) borosilicate glass (1B150F-4; World Precision Instruments, Sarasota, FL) and pull the patch pipettes using a vertical puller (Narishige, PP830; Tokyo, Japan). Open-tip patch pipette resistances in recording solutions are 7-8 MΩ when the pipette is filled with internal pipette solution #1.
- Coat the patch-pipette evenly, from the tip to the level of the shaft that reaches the pipette holder, with dental wax (Cavex, West Chester, PA). This will minimize the pipette capacitance and electrical noise and enable more accurate and low-noise capacitance measurements. A low pipette capacitance also helps the electronic C-fast (fast capacitance) compensation of the EPC-9 (or EPC-10) patch clamp amplifier.
3. Preparation of agar-embedded retinal slices
- Select a goldfish (Carassius auratus; 8-16 cm) and place in a covered bucket in a dark room for 30 min of dark adaptation. After anesthesia, euthanize the sedated goldfish by quick decapitation and double-pithing of the spinal cord and brain stem. Then remove the eyes with curved-tip scissors and curved-tip tweezers. These procedures have been approved by the Institutional Animal Care and Use Committee (IACUC) at the Oregon Health & Science University.
- Hemisect each eye by cutting evenly around the front of the eye with spring scissors (15003-08, Fine Science Tools; initiate cut by puncturing with scissor tips), and place the eyecups in chilled slice solution (low calcium). If necessary, remove the lens with tweezers (the lens will usually detach with the front of the eye). Remove the retina with pigment epithelium attached by using a pair of 45° angled tip fine forceps (11251-35, Fine Science Tools) to peel the retina away from the eyecup, progressing slowly around the full 360°. Sever or cut the optic nerve with either fine forceps or spring scissors. Remove the retina gently with a combination of angled fine tip forceps and suction applied from a modified Pasteur pipette or transfer pipette (large tip). Use angled fine tip forceps to remove the remainder of the pigment epithelium attached to the retina. The surface of the cleaned and healthy retina should appear dark, smooth and red colored. For complete removal of all dark pigment epithelium cells dark-adapt the retina for 1 hour (8, 1992).
- Treat isolated retina with ~0.03% (wt/vol; ~0.36 mg/mL in 1x slice solution) hyaluronidase (3; H6254, Sigma) for 15-30 min at room temperature (20-23°C) to remove vitreous humor. During this incubation, you can prepare ice-cold slice solution.
- Cut a rectangular piece of retina (~2×2 mm, containing the full thickness of the retina) by removing the curved edges with a small segment of razor blade (Personna, double-edged, cleaned with 70% ethanol and H2O) attached to the body of a 1 mL plastic syringe, and transfer the retinal piece to a small container filled with agar solution (prepared in (1.2)). Try to minimize the amount of solution around the retina as it is placed into the liquid agar. Immerse directly in ice-cold slice solution (prepared in (3.3)) to solidify the agar 20, 18, 9. We use a small, handmade container. Briefly, a small, cylindrical tube (Fisherbrand, Polyethylene sample vials 2.5 ml, 03-338-1B) was cut at both ends and sealed with Parafilm (PM-996, Pechiney Plastic Packaging) patches.
- Cut the solid agar block into a 1x1x1 cm cube containing the rectangular piece of retina.
- Transfer the block to the slice chamber and glue it to the surface of the slicing plate. The slice chamber is pre-cooled in a freezer (-20°C). Cut the block into 200-250 μm thick slices using a Vibratome slicer (VT1000S or VT 1200S, Leica) in ice-cold slice solution. A total of 5 to10 slices can be obtained, which are viable for about 5 to 6 hours.
- Transfer one of the slices to the recording chamber. Place a grid of nylon threads glued to a U-shaped platinum frame 19 on top of the slice, and perfuse continuously (rate of 1-2 mL per minute) with recording solution bubbled with 95% O2 / 5% CO2 (carbogen).
Trouble Shooting:
If the retinal piece embedded in the agar block is detached or comes out of the agar during slicing, one can increase the slice thickness from 200 μm up to 300 μm in increments of 20 μm. Also try to minimize the amount of solution around the retina as it is transferred and placed into the liquid agar by sucking up extra solution with a rolled “Kimwipe” paper tip. Another way to avoid this problem is to reduce the size of retinal piece initially placed in the agar. Because vitreous humor prohibits agar from fully attaching to the retinal piece, it may be necessary to make fresh hyaluronidase or adjust the incubation time (step (3.3)).
4. Identification of the bipolar cell synaptic terminal in a retinal slice
- Position the retinal slice under the upright microscope (e.g. BX51WI, Olympus). We view the retinal slice with infrared differential interference contrast (IR-DIC) optics through a 60x water-immersion objective (NA 0.90, Olympus) and CCD camera (XC-75, Sony). The output of the CCD camera is sent to a camera controller (C2400, Hamamatsu) for contrast enhancement before visualization on an analog Sony 13″ black and white monitor. The microscope is mounted on an X-Y translation stage, and the recording chamber is placed on a fixed stage 14.
- Find either intact and axotomized (axon-cut) bipolar cell terminals, which can be identified by their size, shape, and position in the slice (Figure 1). Axotomized and intact terminals can also be distinguished by examination of their capacitative transient current decay times (single exponential versus double-exponential decays, respectively) and Ca2+ currents (Figure 2; 12, 14, 15). Mb terminals are located in the most proximal layer of sublamina b (ON-type layer; adjacent to ganglion cell layer) in the inner plexiform layer of the goldfish retina. Mb terminals, with their dull surface and flat appearance, can be easily distinguished from ganglion and displaced amacrine somas, which appear phase-bright and spherical. Furthermore, the edge of Mb terminals is covered with a high density of synaptic contacts, and appears rough and irregular when compared to the smooth, clean surfaces of ganglion and amacrine cells (Figure 1a-c). Axotomized Mb terminals appear round and near circular (Figure 1c), while intact terminals appear elliptical and stretched, often with a pinched end pointing toward the inner nuclear layer of the retina (Figure 1a,b).
- Damaged or unhealthy terminals often look swollen and display several small granular structures on the surface. It may be possible to obtain a GΩ seal on these terminals, but they are likely to rupture shortly after break-in. Other signs of unhealthy terminals include a hollow appearance, contrast and/or brightness identical to the surrounding slice, and sometimes location at the surface of the slice. It is possible to record both from healthy terminals deep in the slice (advantage: more intact synaptic circuitry) and also near the surface of the slice (advantage: quicker access to perfused drugs in the external solution; easier to seal).
5. Electrophysiological recordings and Ca2+ imaging
- Using a syringe with a 0.2 μm filter tip (Nalgene), fill the glass patch pipette with internal solution to a level 1-2 centimeters from the back of the pipette. After removing air bubbles from the tip, secure the pipette in the holder, apply positive pressure (1.2-1.6 psi), and move it toward the targeted terminal using a micromanipulator (MPC-200, Sutter Instrument). While moving the tip downward through the slice, move the pipette in a lateral sweeping direction to ensure that the slice is not pulled along with the tip.
- Move the pipette tip around the terminal to clean the membrane surface. Push the tip downward on the edge of the terminal to create a dimple (indent), and release the positive pressure. If a GΩ seal does not form immediately, apply slight negative pressure.
- After a GΩ seal, switch to the on-cell recording mode of the EPC-9 patch clamp amplifier and change the holding potential to -60 or -70 mV. Apply the C-fast (fast-capacitance) correction, wait for the seal to stabilize between 5-10 GΩ, and then rupture the membrane with sharp, gentle suction applied by mouth to establish the whole-cell recording configuration. If the whole-cell configuration has been correctly established, series resistance will be between 14-30 MΩ. Input resistance will be 200-500 MΩ for intact terminals and 1-3 GΩ for axotomized terminals 14.
- Observe the capacitive current transient (Figure 2a and 2c), to distinguish between intact and axotomized bipolar cell terminals. Intact and axotomized Mb terminals have a baseline membrane capacitance of 9-16 pF and 3-8 pF, respectively.
- Apply a 200 ms duration voltage-clamp step from -70 to 0 mV to an axotomized Mb terminal. This will allow the direct measurement of large (~200-600 pA), isolated inward Ca2+ currents activated by the opening of voltage-dependent L-type Ca2+ channels (Figure 2b). In parallel, one is able to monitor the capacitance jump caused by the exocytosis of synaptic vesicles, and the rapid, pH- mediated inhibition of Ca2+ current that follows exocytosis 15, and GABAergic reciprocal feedback inhibition (Figure 3c; 22). If one patches an intact Mb bipolar cell terminal the result will be as shown in Figure 2c and 2d. No discernible Ca2+ currents are observed because of the large “leak” currents due to gap-junction coupling in the dendrites of the Mb bipolar cells 1.
- Membrane capacitance changes can be evoked by a step depolarization from the holding potential of -70 mV to 0 mV using the “sine + DC” method of time-resolved membrane capacitance measurements 6. Membrane capacitance jumps indicate the fusion of synaptic vesicles with the plasma membrane. A 2 kHz sinusoidal voltage-clamp command (30 mV peak-to-peak) was added to the holding potential of -70 mV. The recording current was analyzed at two orthogonal phase angles by the EPC-9 patch-clamp amplifier software emulation of a lock-in amplifier (HEKA, Lambrecht, Germany). In the intact terminal of Figure 2d, a high frequency (2 kHz) sinusoidal stimulus confines the membrane capacitance that is analyzed to the terminal endings and axon, which have a baseline capacitance Cm ~6.8 pF. The membrane of the cell body and dendrites of the bipolar cell are thus filtered out by the high frequency voltage-clamp sinewave 13.
- For Ca2+-imaging experiments, thoroughly mix the internal patch pipette solution with a Ca2+-sensitive fluorescent dye (e.g. Oregon Green 488 BAPTA-1 (100-200 μM; O-6806, Invitrogen)) and repeat the protocol from 5.1 to 5.5. This will allow one to combine fluorescence imaging and electrophysiological recording. For example, it is possible to image the Ca2+ transient associated with a 200 ms step from -70 to 0 mV in the small telodendria appendages of the Mb terminal (Figure 3a,b). We have integrated our upright Olympus microscope with a spinning disk laser confocal microscope system (CSU-X1, Yokogawa). The confocal microscope uses 488 nm and 561 nm laser lines that are modulated by an acousto-optic tunable filter. Data are acquired using Slidebook software (3i; Intelligent Imaging Instruments). For further details on calcium imaging techniques using goldfish retinal neurons see 24, 17, 3.
Trouble Shooting:
If one frequently fails to establish a whole-cell configuration, even after formation of a stable GΩ seal, it may be helpful to reduce the negative pressure used to puncture the terminal membrane. It may also be the case that optimal suction pressure for the establishment of whole-cell configuration varies as a function of membrane curvature (soma > terminal). It is also possible to use a zap protocol (Amplitude: 400 mV, Duration: 100 μs) to enter the whole-cell configuration, either alone or in combination with a sharp pulse of negative pressure. We have found the zap protocol to be particularly useful for break-in when using a pipette tip with a bath resistance in excess of 12 MΩ.
6. Representative Results
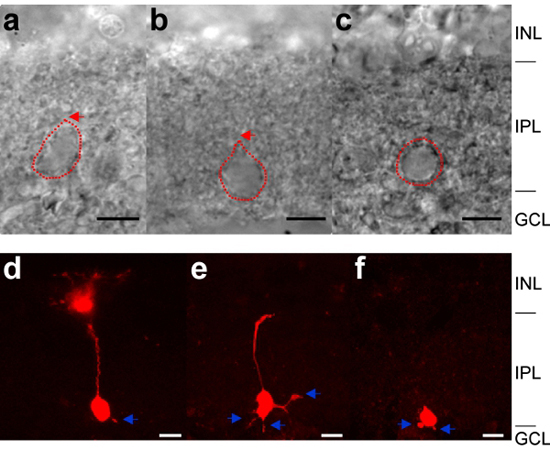
Figure 1. Identification of Mb bipolar cell terminals in goldfish retinal slices. (a-c) IR-DIC images of Mb bipolar cell terminals. We can identify likely axotomized (axon-cut) and intact terminals in the IR-DIC image. An axotomized terminal appears round and circular, as shown in c while an intact terminal is more likely to appear elliptical, as shown in a and b. This classification can be confirmed by transient capacitance measurement (see Figure 2) or by the dye filling method (d–f). Red arrows indicated the location of the intersection between the axon and axon terminal for the intact terminals in a and b. Red dotted line indicated the contour of Mb bipolar cell terminals. (d-f). Fluorescence images of Mb bipolar cell terminals with an intact axon (d), a partially cut axon (e), or a fully removed axon (f). Alexa 555 (A20501MP, Invitrogen) fluorescent dye was filled with internal solution prior to recording. Immediately after patch-clamp recording, the retinal slice was transferred into 4% (wt/vol) paraformaldehyde (P6148, Sigma) in phosphate buffer solution (70013, GIBCO) and incubated for 30 min. Slices were mounted onto Superfrost slides (Fisher Scientific) in aqueous mounting medium with anti-fading agents (Biomeda corp). Alexa 555 containing Mb bipolar cell terminals were viewed with a 555 nm laser line (red) using a 40x water-immersion objective on a confocal laser-scanning microscope (LSM 710, Carl Zeiss). Note the presence of small telodendria appendages protruding from the axon terminals (blue arrows). Scale bar indicate 10 μm (a–f). INL: inner nuclear layer, IPL: inner plexiform layer, GCL: ganglion cell layer.
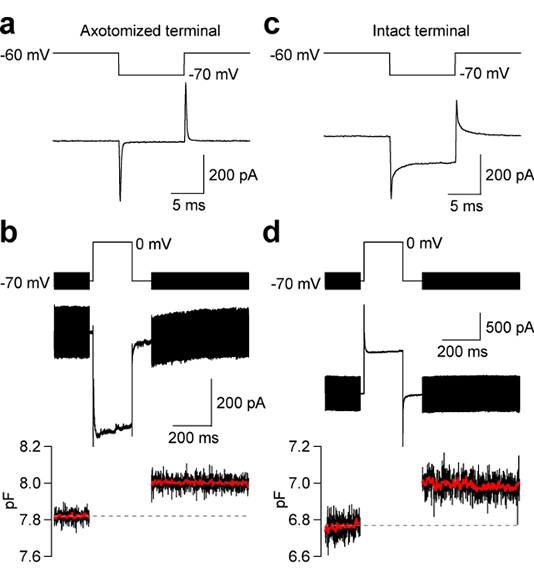
Figure 2. Electrical properties of axotomized and intact Mb bipolar cell terminals. (a,c) Transient current response activated by a voltage-clamp step hyperpolarization from the holding potential of -60 mV to -70 mV. The short current response to a -10 mV voltage-clamp step can result in: 1) a single fast exponential current decay with high input resistance at -70 mV (indicative of an axotomized terminal; panel a), or 2) a double-exponential decay with low input resistance at -70 mV (indicative of an intact terminal; panel c; see also 12, 14, 15). (b,d) Ca2+ currents and membrane capacitance jumps were evoked by a step depolarization from -70 to 0 mV. Time-resolved membrane capacitance measurements use a 2-kHz sinewave stimulus superimposed on the resting holding potential of -70 mV 6. The red trace is the averaged value of the capacitance data points. Note that the capacitance jump in Figure 2b and 2d are both equal to about 200 fF.
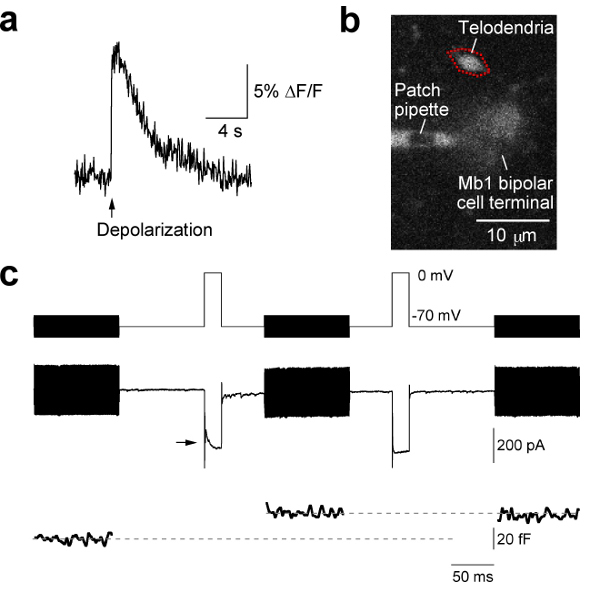
Figure 3. Direct measurements of Ca2+ imaging signals, exocytosis, and inhibitory feedback in an Mb bipolar cell terminal. (a) A transient rise in Ca2+ concentration in the telodendria of an Mb terminal was activated by a voltage-clamp step depolarization from -70 to 0 mV for 200 ms (arrow). Oregon Green 488 BAPTA-1 (100 μM), a Ca2+ indicator dye, was included in the patch pipette. (b) Corresponding fluorescence image of the Mb telodendria (region of interest indicated by red dotted line). (c) Short-term synaptic depression of neurotransmitter release (exocytosis). Ca2+ currents (middle trace) and membrane capacitance (lower trace) evoked by a pair of 20 ms depolarizations to 0 mV (upper trace; 200 ms inter-pulse interval). The arrow indicates the effect of exocytosed protons on the Ca2+ current and also the GABAergic reciprocal feedback inhibition from neighboring amacrine cells 15, 21. Note that the proton-mediated inhibition of the Ca2+ current and also the GABAergic reciprocal feedback inhibition are present only in the first depolarizing response, which also has a capacitance jump due to the exocytosis of synaptic vesicles.
