1. Optical Calibration: Measuring LED Power
- Attach a LED array to a heatsink actively cooled by a fan and affix this LED/heatsink aparatus to a collimating lens.
- Replace lamp used in brightfield illumination with LED/heatsink/fan/lens apparatus. This apparatus must be carefully positioned so that the collimated LED beam travels along a straight optical path towards the condenser lens. Make sure the heatsink/fan is properly grounded to the system’s common ground.
- Drive the LED array with a power supply that can give fast and square pulses of current. This power supply may be controlled by a 5V TTL pulse originating from a pulse generator.
- Center the collimated beam along the light path defined between the field diaphragm and the condensor lens. Ideally, the LED beam will slightly overfill the fully opened field diaphragm. Typically, a more tightly collimated LED beam will fill this aperture less, and will produce more power at the expense of uniformity. In our setup we increased light uniformity by choosing a collimating lens that projected a slightly expanded image of the LED array at its conjugate plane at the consenser diaphragm.
- Achieve Kohler illumination by focusing the condenser so that the image of the field diaphragm (the diaphragm closest to the light source) is focused on the slice chamber (fig. 1). A thin tissue of lens paper can act as a projection screen to visualize the focused image of the field diaphragm at other depths.
- Drill a series of pinholes of known diameter in an opaque material. Place one of these small pinholes over the sensor of an optical power-meter. Place the power meter on the specimen stage and center the power meter over the focused image of the field diaphragm by simply moving the power meter until it gives a maximum reading. Affix the power meter in this position.
- Fully open all apertures (aperature diaphragm and field diaphragm). Systematically move the attached slice chamber/power meter relative to the optical lightpath and calculate the uniformity of optical power within the illuminated area. Build the uniformity plot for your system. If the microscope is properly setup with Kohler illumination centered at the objective’s focus, maximum power should be directly underneath the objective, and regions outside of this focus should now receive a known amount of power according to the uniformity plot.
- For each size of pinhole, build a standard curve of optical power vs. pinhole surface area. By adjusting the input current to the LED array, produce this curve at multiple power levels and from each curve calculate the power per mm2. If the LED array is to be used for patching optics, make sure to introduce optical elements necessary for patching (condensers, pinholes and filters) to know how much light is transmitted under patching illumination.
- Flipping the power meter to face the objective, calculate the intensity of illumination at 470nm when the mercury lamp is turned on.
- Add a live slice to the chamber, and rebuild standard curves to determine the light power transmitted through the scattering brain tissue.
2. Slicing Procedure and Electrophysiology
Part A: Slice Preparation
- Anesthetize (60 mg/kg Ketamine and 2mg/kg Xylazine) and decapitate the mouse. Dissect the brain in artifical cerebral spinal fluid (ACSF, in mM: 124 NaCl, 3 KCl, 1.3 MgSO4, 26 NaHCO3, 1.25 NaHPO4, 20 glucose, 2 CaCl2; ~310mOsm, pH 7.4 when bubbled with a mixture of 95% O2 and 5% CO25,1), taking care not to damage the olfactory bulbs. Separate the two hemispheres and place on agar with the ventral surface even with one edge (for horizontal sections).
- Glue the agar and dorsal surface of each cortical hemisphere to the vibratome chuck, and slowly fill the bath with ice-cold ACSF. Slice from ventral surface in 300 μm sections, transferring each section to warmed (34-36°C) and oxygenated ACSF, allowing them to recover for 30-45 minutes.
Part B: Loose Patch Measurement of the Threshold to Spike
- After bringing the slices to room temperature for 30 minutes, gently place a slice in the recording chamber of the microscope chamber under constant perfusion of oxygenated ACSF.
At risk of excessively stimulating ChR2 infected neurons, the presence of EYFP-ChR2 can be confirmed under epifluorescence (fig. 2a) - Pull glass electrodes on a pipette puller (Sutter P-97). Fill this electrode with ACSF. When placed in the ACSF bath the tip resistance should be between 7-10 MOhms.
- Under fluorescent illumination, locate in the slice a healthy ChR2-EYFP neuron with mature morphology. Also locate this neuron’s soma under patching optics.
- With light positive pressure passed through the electrode tip, lower the patch electrode towards the identified fluorescent neuron. When membrane contact is made, quickly release positive pressure and apply a small and brief amount of suction through the tip. A giga-ohm seal should be made between the plasma membrane and the walls of the patch electrode.
- Even if a giga-seal is not formed, if the electrode is sufficiently close to a fluorescent neuron spiking activity should produce a measurable local field potential. Activate ChR2 in this neuron by flashing different doses of light. Because light-dose is a function of both LED power and duration, calculate how much light is necessary to evoke an action potential at multiple powers and durations (fig. 2b). Also observe how much spiking occurs under mercury lamp illumination.
3. Representative Results:
On our microscope (Olympus BX51WI), our LED is in line with 2 apertures and a condenser lens, thus maintaining the original lightpath of the factory-installed arc lamp. By closing both the field diaphragm and the aperature diaphragm, we can achieve brightfield contrast sufficient for patch clamp recordings (fig. 1b). With all diaphragms fully open we expose the slice to maximum light power for channelrhodopsin activation (fig. 1a). On our microscope, this patching configuration produces light-density that is approximately three orders of magnitude lower than the maximum full field density (4.1 μW/mm2 versus 6.88 mW/mm2).
We see robust labeling of adult-born olfactory bulb granule and periglomerular neurons weeks after lentiviral infection of migrating neuroblasts in the rostral migratory stream (fig. 2a) A loose-patch recording from a single adult-born ChR2-EYFP expressing granule cell indicates that a 5 ms stimulation at maximum power (6.88 mW/mm2) is sufficient to evoke spiking (fig. 2b). Since expression level varies between cells, the amount of light that passes the threshold to spike will vary and should be described statistically for each cell type of interest.
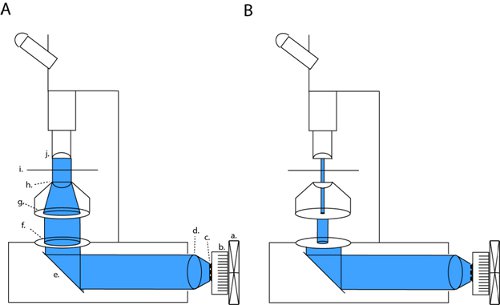
Figure 1. LED array setup for full-field photostimulation and patch-clamp slice electrophysiology. To activate channelrhodopsin (ChR2) we project a collimated beam through open back apertures and condenser optics (a). This configuration can be changed into high-contrast patching optics by fully closing the field diaphragm and modulating the width of the field diaphragm (b). Abbreviations: a. fan, b. heatsink, c. LED array, d. collimating lens, e. mirror, f. field diaphragm, g. aperture diaphragm, h. condenser lens, i. sample stage, j. objective.
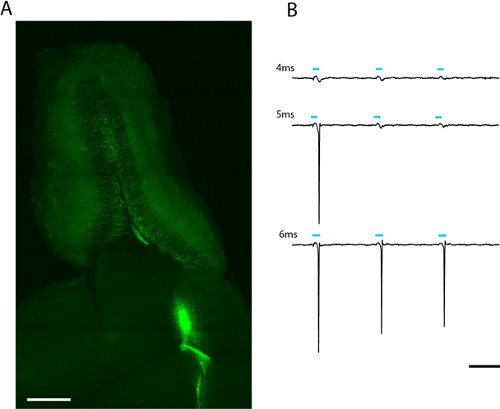
Figure 2. Image of a 300μm horizontal slice of olfactory bulb for patch clamp and whole-field photostimulation (a). Lentivirally infected adult-born granule cells expressing ChR2-EYFP can be seen radiating from the core of the olfactory bulb. The light-dose required to evoke spiking can be found by increasing the duration of the LED flash (b). The threshold for this granule cell was 5ms at full LED intensity (2.43mW/mm2). Scale in (a) = 500μm, scale in (b) = 50ms.
