The protocol is adjusted for the purpose of statistical analysis of MALDI IMS data from multiple rat brain sections, typically 20-30 sections, and consists of five different steps comprising tissue preparation, matrix application, MALDI-TOF MS analysis, data evaluation, and neuropeptide identification. The procedures are outlined and described in more detailed below:
1. Tissue preparation
This procedure includes the collection of the respective tissue samples as well as tissue sectioning for IMS analysis. A particular objective in protein and peptide analysis is to avoid proteolytic degradation. Therefore it is essential to work fast and diligent during tissue dissection.
- Sacrifice rats (typically 250-300 g) by decapitation, remove rat brain within a maximum post-mortem time of <30s and freeze on powdered dry ice before transferring to -80°C freezer. Faster freezing using liquid nitrogen increase the risk of microtears in brain tissue, which will negatively affect matrix crystallization and thereby reduce MS quality (Fig. 2D). Whole brains can be stored for several years before sectioning without loss of MS signal quality.
- Cut frozen tissue on a cryostat microtome to 12 μm slices and thaw-mount tissue sections on conductive MALDI glass slides (indium tin oxide coated slides, Bruker Daltonics) or MALDI target (Fig. 2A-C).
- Dry sections for 15 min under vacuum and store slides at -80°C until further use. Tissue sections should be analyzed within the shortest possible time after sectioning, even if stored at -80°C. We find that MS signal quality will be noticeably reduced after a year in storage. In order to reduce oxidation of proteins and peptides, the air in storage container can be replaced with an inert gas (e.g. argon or nitrogen).
2. Matrix application
The matrix application step has a significant impact on spectrum quality and requires optimization of multiple parameters depending on the kind of tissue as well as the analyte of interest. These factors include chemical parameters such as the kind of matrix, matrix concentration, pH, tissue washing and organic modifiers as well as instrumental settings including the deposit volume, lateral resolution and number of depositions10 (Fig. 2D). For large scale experiments, it is of great importance to reduce variance, for example by applying the matrix to all sections within one day and by the same operator. Although there are many strategies to apply matrix solution such as by sublimation or by spray, the automated deposition of arrays of small matrix droplets, about 100-150 picoliter in size, has been used successfully for the analysis of small proteins and neuropeptides in various tissues, including brain sections9, 10,11, 12, 13.
- Defrost the sections in a dessicator for 1 hour.
- Make sure that the experiment is blinded by a person other than the operator. Re-label all samples.
- Wash sections 1x in 70% ethanol (EtOH, at room temperature, RT) for 10 sec and twice in 95% EtOH (RT) for 10 sec. For large experiments, perform the washing for all glass slides together using a cuvette in order to minimize variation.
- Dry the sections in a dessicator for 10 min.
- Evaluate tissue sections under a microscope and check for tissue distortion, microtears and small cracks that will impair MALDI MS quality (Fig. 2D).
- Prepare fresh matrix solution consisting of 50 mg/mL DHB in 50% methanol, 10% 150mM ammonium acetate (AmAc) and 0.3% trifluoroacetic acid (TFA) in water.
- Matrix application is performed by discrete droplet deposition in a rectangular pattern using a chemical inkjet printer (CHIP, Shimadzu). The first step is to optimize the experimental parameters of matrix application for neuropeptide analysis including the number of droplets per pass, number of passes. This experiment is performed by applying multiple matrix arrays with different application parameters on the same tissue section, while making sure that each array is covering similar brain regions such as corpus callosum, cortex, and striatum. The same experiment has to be performed each time parameters are changed, including different brain structures, different matrices targeting specific analytes, and if different matrix solvents are needed for extraction of specific analytes.
- Scan the glass slide holder with the tissue section and align the holder. Define your array for matrix application on the tissue section and specify the spatial resolution i.e. spot to spot distance. Apply matrix using the optimized protocol on the chemical inkjet printer. For this experiment we used an optimized protocol for peptide imaging with the following printing parameters: 10 drops (100 pL/drop), 10 application passes and a spot to spot distance of 300 μm.
- Scan final matrix spotted sections and save the picture for registration prior to MALDI data acquisition (step 3.4).
- Store the sections until further use in a dessicator under vacuum.
3. MALDI MS data acquisition and processing
MS analysis of neuropeptides is performed on a MALDI time of flight instrument (Ultraflex II, Bruker Daltonics, Germany) operating in reflector mode, using software assisted data acquisition from every single matrix spot14. Therefore accurate spatial teaching is imperative. It is essential that the MALDI optimization, acquisition and especially the target registration experiments are performed by the same operator that should preferably be blinded to the experimental groups. In a large scale experiment with multiple glass slides, the MALDI experiments can be performed by one operator while another person is operating the chemical inkjet printer.
- Load glass slides into the mass spectrometer.
- Check the calibration of the MALDI acquisition method using a low molecular weight standard calibration mix (Bruker Daltonics).
- Optimize acquisition parameters.
- In order to optimize MS signal and to avoid ablating matrix from neighboring matrix deposits, the size of the laser and the optimum focus on tissue should be determined.
- The laser energy is set to ensure the maximum MS quality from as many matrix deposits as possible without elevating the baseline, reducing peak resolution or saturating the detector.
- Assess the maximum number of shots per matrix spot until only noise is detected, often 1000-2000 shots. Estimate the number of shots that should be accumulated and the number of shots acquired before the laser position within a spot should change. In order to sample each matrix spot evenly, we accumulate 600 shots in 25 shot steps, for a total number of 24 steps using a random pattern of movement, from each matrix deposition.
- Register the scan of all spotted sections to motor coordinates of the MALDI stage using the FlexImaging (v.2.0)10 and perform data acquisition in batch mode by AutoXexuteBatchRunner.exe software.
- Process each single spectra by means of baseline subtraction (Convex hull V3), smoothing and external calibration (optional), followed by export as an ascii file (*.dat, *.txt or *.csv format).15
4. Data evaluation
Final data evaluation comprises data post processing and data reduction by focus only on peak information, followed by statistical analysis.
- As an initial step, the MALDI IMS sections were evaluated for overnormalization effects. This can be easily achieved by employing data visualization tools such as FlexImaging (Bruker Daltonics) or BioMap (Novartis). As an initial step the total ion images are evaluated before total ion current (TIC) normalization, followed by manual inspection of single ion distribution images of various prominent peptide peaks. Look for characteristic peak intensity distributions and if they are related to tissue features (damages), spotting quality or normalization effects (Fig. 3).
- Delineate regions of interests (e.g. the striatum) according to histological features and export the corresponding spectra in ascii file format. Preferably, normalization of spectra to the total ion current (TIC) can be performed at this stage.
- Import ascii files in a data handling software such as Origin (v.8.1, Originlab), MATLAB (MathWorks, Natick, MA, USA) or R16. Peak detection can be performed using peak finding tools included in the software, for example “peak analysis” in Origin or “mspeaks” in Matlab. Export the peaklists from all spectra as one single tab delimited text file.
- In order to determine bin borders for detected peptide peaks, binning analysis is performed using appropriate software tools (e.g. pbin17) or in-house written scripts for MATLAB or R. Here the single text file containing all peak picked data is loaded into the software and the parameters for peak border determination are specified such as how often a peak should be present in spectra in order to be relevant for the experiment. For example, the experiment contains 2 groups of animals, 5 animals in each group, and 100 spectra are collected from each animal and region of interest. Assume a peak is potentially interesting if it is present in at least the majority of the animals in one group (3/5) and in at least half of the spectra of those animals (3×50=150 spectra), this will give a total percentage of 15% for the 150 positive spectra out of the total 1000 (2x5x100) spectra. Using the pbin tool, this step yields a single binrange file containing all bin widths determined from the acquired data. In order to verify that bin borders are appropriate, it is easy to visualize the bins in Origin together with the original spectra traces.
- Peak-area integration can reduce variance that is important for statistical analysis. We use an in-house written script for R to calculate the area under the curve between peak borders determined in step 4. Integrated peak areas are imported into MS excel (v.2007) and statistical analysis by means of non-parametric unpaired testing using the SAM tool is performed18.
5. Peptide identification
Sequence verification of the observed peptide identities is essential in order to conclude biological relevance. The most accurate approach include true top-down determination directly off tissue using peptide fragmentation by means of tandem mass spectrometry (MS/MS), although high peptide concentrations are required for this kind of analysis12,13. For low abundant peptides or multiple peptides with close m/z values (±0.5%), on-tissue analysis is impaired and off tissue analysis using a peptidomic strategy is utilized that includes extraction, separation and MS based identification of endogenous neuropeptides. For the experiment presented here, the central focus was on opioid peptide detection, which is a particular challenge since these peptides are quite low abundant compared with other neuropeptides in the spectra. Furthermore, these peptides are rather polar which makes them relatively hydrophilic and difficult to retain with common peptide extraction and separation techniques.. Therefore we applied a previously reported protocol for tissue extraction and opioid peptide prefractionation in combination with standard LC-MS/MS based peptide identification9,19.
- Collect coronal sections of target structures of interest (nucleus accumbens, NAc; caudate putamen, CPu). Mount frozen rat brain in a cryostat microtome and remove surrounding brain material (cortex, septum, corpus callosum) with a scalpel. Collect sections (30 μm; n=50) of the dissected NAc and CPu and thaw mount the NAc and CPu parts of the sections on different glass slides.
- Extract peptides off tissue by adding 100 μL 5%ACN/0.1% TFA, incubate for two minutes and collect in eppendorf low protein binding tubes. Repeat this step twice.
- Perform peptide prefractionation by means of strong cation exchange chromatography using stepwise (n=4) elution at increased ionic strength19. Dry down samples under vacuum using a speedvac concentrator.
- Analyze the peptide fractions by means of nanoflow C18 reversed phase liquid chromatography (1100, Agilent Technologies, Santa Clara, CA) interfaced to electrospray tandem mass spectrometry (LC-MS/MS). The MS experiments were performed on a hybrid linear iontrap/Fourier transform ion cyclotron resonance (FTICR) instrument (LTQ FT 7T, Thermo Scientific, Waltham, MA,) Peptide fulscan spectra (m/z 150-2000) were acquired with the FTICR analyzer at high mass resolution (100000) followed by subsequent fragmentation of the 5 most intense peptide peaks in the iontrap by means of collision induced dissociation (CID)9.
- Peptide sequence identification is performed by database matching and can be complemented by de-novo sequencing analysis. For database search, commercially available search engines (Mascot, XTandem or Protein prospector) are employed20. The searches are typically performed against databases containing sequences of known or predicted neuropeptides and sequences of neuropeptide precursor proteins21.
6. Representative Results
MALDI imaging mass spectrometry of striatal tissue sections as prepared according to the protocol described here resulted in the detection of more than 1000 peaks corresponding to approximately 300 monoisotopic molecular species (average spectra shown in Fig. 1). Data visualization for prominent molecular ion peaks was achieved using the Flex Imaging software and showed characteristic peak intensity distributions that are well in line with anatomical features (Fig.3). A further feature of MALDI IMS is its relative good reproducibility. In this experiment, the overall coefficient of variance for the peak intensities of all detected molecular species was 30%, but many peaks displayed very low variation and high reproducibility within treatment groups (Fig. 4). The relative peak intensity data of four different regions of interest, including the dorsolateral and dorsomedial part of both lesioned and intact striata were subjected to statistical analysis. In order to adjust for multiple comparisons simultaneously, the statistical analysis was performed by means of nonparametric testing using the SAM tool18. The most prominent changes were found in the dorsolateral part of the dopamine-denervated, parkinsonian striatum. Here significant changes in between the different treatment groups were observed for two dynorphin peptides; dynorphin B and alpha-neoendorphin (Fig.5). In detail, a relative increase of both dynorphin peak intensities by 50-60% was observed in high dyskinetic animals compared to low dyskinetic animals and lesion controls (p<0.05, F(2, 15) = 12.8 DynB, F=5.7 aNeo; Fig. 5).
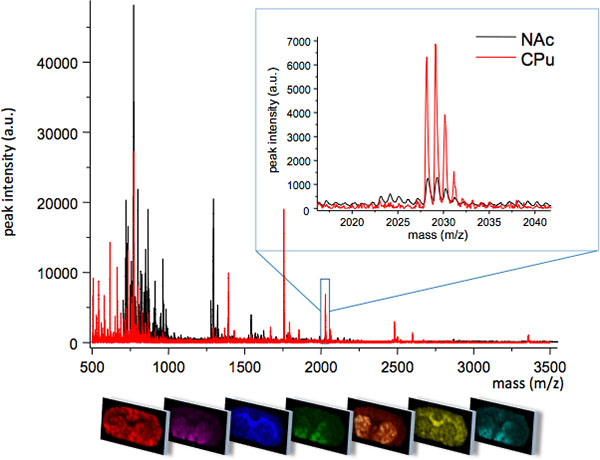
Figure 1. Average MS traces obtained from two closely related regions of the striatum; caudate putamen (CPu) and nucleus accumbens (NAc). The two regions display different MS profiles with some molecular species uniquely expressed in one region, or at different peak intensity levels (insert, m/z 2028). The spatial distribution pattern of each peak can be visualized using specialized imaging software (bottom panel).

Figure 2. (A) The brain is mounted on a cryostat chuck using an embedding media (OTC; arrow), care is taken that the OTC does not contaminate the area of brain to be sectioned since the OTC cause ion suppression of peptides. (B,C) Thin sections (≈12 μm thickness) are thaw-mounted onto MALDI compatible glass slides and dried for a few seconds to avoid freeze damage as seen in C. (D) Microtears may be difficult to detect to the naked eye, but impair MALDI matrix crystallization and obliterate MALDI MS signal. The same section stained with cresyl violet reveals microtears and cracks (bottom right photomicrograph).
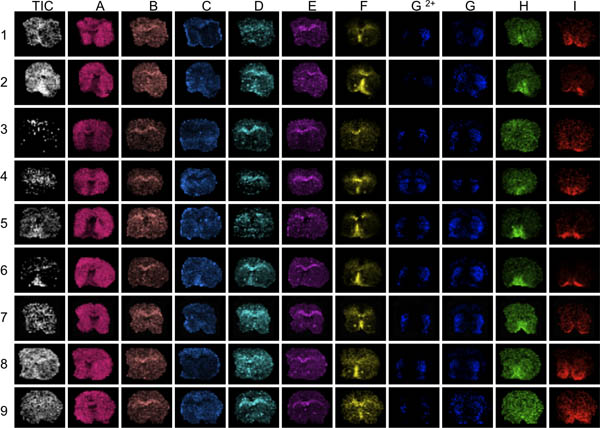
Figure 3. The first step in data evaluation is to visualize several different peaks across the mass range analyzed (A-I). Here, striatal sections from 9 mice were imaged with MALDI MS. Visualization of the average total ion current will reveal areas of conspicuous high or low ion intensities (arrows). These areas can be affected by over- or under-normalization effects and distort data analysis compromising the results. Poor anatomical definition of peak distributions reveal sections with generally low peak signal-to-noise, for example sections 3 and 9, peaks F through I.
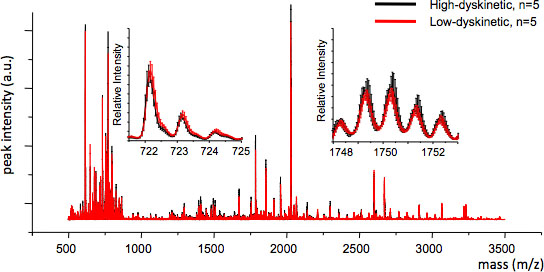
Figure 4. MS reproducibility between treatment groups can be assessed by calculating the average MS trace and the standard error for each m/z value (inserts, m/z 722 and 1749). Good reproducibility ensures valid statistical analysis.
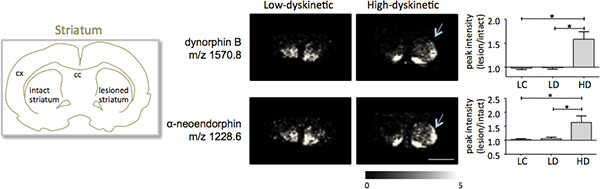
Figure 5. Dynorphin B and alpha-neoendorphin peak intensities are significantly increased in the 6-OHDA-lesioned, parkinsonian, striatum of high-dyskinetic animals (HD; arrows) compared to the low-dyskinetic (LD) and lesion control group (LC). Peptide peak intensities expressed as average fold-change of intact side ± SEM (lesion/intact side). * p<0.05; cx cortex; cc corpus callosum. Scale bar 5 mm. Please click here to see a larger version of this figure.

{kind=link}