1. Preparing the animal for imaging
Note: We use adult c57BL mice. Different transgenic strains can be used depending on the research question. Microvascular surgeries and pulse oximetry are facilitated in mice with white fur (e.g. FVB strain).
- Prepare the surgical site at the bench. All surgical instruments need to be autoclaved or disinfected with 70% ethanol.
- Insert a rectal probe, and continuously measure the animal’s body temperature throughout the procedure
- Anesthetize the mouse using initially 5% isoflurane. In 15-30 s, when the animal is not moving, place it on a heating pad (37 °C), and apply a face mask for the continuous delivery of air containing 1.3-1.5% isoflurane). Make sure that the animal is not responding to a pain stimulus (e.g. tail pinch).
- Use artificial tear gel to cover the mouse’s eyes, to prevent exposure to dry air.
- Using an electric razor, remove hair from the head and from both thighs. Apply hair remover (e.g. Nair) to the thigh for 2 min, and then wipe it carefully to remove the remaining hair. This thigh will be used for oximetry.
- Disinfect the scalp using a 10% povidone-iodine solution and 70% ethanol.
2. Preparing the open skull cranial window
- Starting 5 mm caudal to the skull, make an incision in the scalp with scissors, and advance one centimeter. Move skin to the sides to expose the skull.
- To remove the membranes on top of the skull apply 10% ferric chloride solution to the top of the skull. Wipe away the excess solution, and scrape the membranes away with 45° angle #5 tweezers. It is important to prepare the site carefully, in order to ensure that the head plate can be securely attached to the skull (steps #4 thru 6).
- Using a dissection microscope, identify the area of interest. Note that the cranial sutures should be avoided because the skull can break unpredictably along these lines.
- Apply a thin layer of rapid glue (e.g. Locktite 454) around the edges of the window of the head plate (Figure 1). Position the head plate in such a way that the area of interest is exposed in the window. Apply light pressure on the head plate. Apply a small amount of dental cement to polymerize the glue rapidly. Hold for 10 seconds while the glue is polymerizing.
- Apply a small amount of rapid glue to the edges of the window to seal the head plate and the skull. Screw the head plate to the animal holder under the dissection scope.
- Using the Microtorque II drill set at 6000 rpm and a IRF 005 drill bit, remove all extra glue from the skull and from the head plate. Any remaining glue on the head plate should be removed, since it will otherwise interfere with the attachment of the glass cover.
- Set the drill at 1000 rpm, and start drilling the skull by making a circle inside the head plate window, diameter ~5 mm. Use light sweeping movements as if you are drawing with a very soft pencil. Stop drilling every 20-30 seconds to remove bone dust using compressed air.
Note: The shape of the opening in the head plate allows additional access for the drill bit (for left- and right-handed surgeon), which makes it easier to create a circular cranial window. In addition, the two odd shaped holes provide the required space to insert a Clark-electrode or glass-electrode into the brain tissue under the cranial window for additional polarographic or electrophysiological measurements. - As the drilling progresses, a burrow is formed around the intact skull in the center. Be careful not to poke through the thinned skull, but to make this burrow of even thickness. Be extra careful around large blood vessels -they should not be compromised and, if possible, not even touched.
- Use one “leg” of tweezers #3 to lift (“flick off”) the bone in the center of the window. Apply a drop of artificial cerebrospinal fluid (aCSF) on the window.
- Wipe aCSF gently using a corner of soft tissue paper (e.g. Kimwipe). Usually the dura is damaged in one or more places around the edge of the window. Starting at one of these sites, gently lift and then remove the dura from the brain surface, using 45° angle tweezers #5. When choosing the site at which to start removing the dura, avoid the areas adjacent to large blood vessels. If bleeding occurs, apply a drop of aCSF and wait for 2-3 min for minor bleeding to stop.
- Prepare 0.07% low-melting agarose dissolved in aCSF) Bring the melted agarose to body temperature. Wipe away the excess of aCSF from the surface of the brain. Pour the agarose in the window, and cover with a glass slide. Press the glass gently to make a contact between the glass and the head plate, and wait 10-20 sec until the agarose is solid.
- Remove excess agarose from the glass cover using tweezers and soft tissue paper.
- Put a small amount of rapid glue around the glass to glue the glass to the head plate. Apply a small amount of dental cement to solidify the glue (Figure 1).
3. Intravenous injection and blood oxygenation monitoring
Note: We routinely used the femoral vein for intravenous application of Texas red rather than the tail vein. This is because femoral vein injections provide safe and reproducible bolus injections of the dye.
- Turn the animal partially over to expose the inside of the left thigh. Use tape to secure the animal in this position.
- Apply antiseptic scrub, and then 100 % ethanol to the skin.
- Make an incision along the medial thigh from the knee to the pubic symphysis. Separate soft tissues by blunt dissection using tweezers #5.
- Fill 1ml syringe with 130 μl of Texas Red (2.5 mg/ml). Attach a new needle (gauge 30), and bend it carefully at 30°, keeping the bevel up. Fill the needle with Texas Red solution.
- Under a dissection microscope, insert the needle into the femoral vein and inject 100 μl of Texas Red. Remove needle and lightly press the vein with gauze to stop bleeding.
- Close the skin using 4-0 suture.
- For continuous monitoring of blood SpO2 (to ensure adequate blood oxygenation) place the oximeter probe on the right thigh (from which hair was previously removed).
4. Two-photon imaging
Note: We use an Olympus Fluoview1000 multiphoton imaging system (upright) with a Spectra-Physics MaiTai HP DeepSee femtosecond Ti:Sa laser as an excitation source. For imaging, we use × 10 NA 0.45 (Zeiss C-Apochromat), or × 25 NA 1.05 (Olympus XLPlan N) water-immersion objectives. We take images at 12-bit depth at a resolution of 512 × 512 pixels with a pixel dwell time of 2 μs. The NADH and Texas-Red-dextran are concurrently excited at 740 nm, and fluorescence is separated from the excitation light using a dichroic mirror/near-IR-blocking filter combination (FF665-Di01; FF01-680/SP, Semrock, Rochester, NY, USA) divided into two channels using a dichroic mirror (505DCXRU, Chroma, Rockingham, VT, USA), and bandpass filtered (NADH-Semrock FF460/80Texas-Red-dextran-Semrock FF607/36). Laser power measured after the objective was 10-20 mW for imaging in layer I and 20-50 mW for imaging in layer II.
- Transfer the surgically prepared mouse to the microscope stage. Take an initial picture of the cranial window site using bright field illumination at 4x magnification to use as a reference map for registration with higher magnification two-photon angiographies.
- Zoom in on the area of interest using × 10 magnification. Select an area of interest in cortical layers I or II (up to 150 μm below the pial surface). If higher magnification is desired, use × 25 objective
- Start record the time series (Figure 2). We recommend using an average of 3-5 frames to increase the quality of the image.
- Induce hypoxemia by adding 50 % N2 to the air that the animal is breathing. This will bring the O2 level to 10 %. Verify hypoxemia by monitoring oximeter data.
- Continue to record the time series through hypoxemia (for example, for 30 seconds), and after normal O2 level is restored.
- Collect data for calculation of the oxygen distribution by detecting, measuring and quantifying the area of hypoxia and oxygenated tissue cylinders around the blood vessels.
5. Data processing
- Determine the Krogh tissue cylinder radius R. This can be done manually (Figure 3) by measuring the distance between center of the blood vessel and the boundary at which high NADH fluorescence is detected.
- Alternatively, use the computational investigator-independent semi-automated determination of the tissue cylinder radius R and the central blood vessel r that has been described previously1 in supplementary form and summarized in the discussion section of this protocol.
- Determine the area of hypoxia using open-access image analysis software, e.g. ImageJ. To do this, use NADH signal, define a threshold that delineates the high NADH intensity area, and then measure the area with elevated NADH tissue fluorescence.
6. Representative Results
Figure 2 Shows a video of NADH/angiography images during transient hypoxemia (for the PDF version of this Figure, selected still images have been used). The inspired oxygen concentration was lowered from 21 % to 10 % for a period of 3 min. This level of experimentally induced hypoxemia is sufficient to induce severe hypoxia in the cerebral cortex. Hypoxia led to an increase in NADH fluorescence, initially in the areas which were furthest from the arterial blood supply. Note the sharp NADH tissue boundaries, which represent observable tissue boundaries of oxygen diffusion from the cortical microcirculation. The image in Figure 3 shows the geometry of oxygen diffusion boundaries surrounding cerebral vessels. It is possible to deduce Krogh cylinder diameters from these boundaries, as described previously 1.
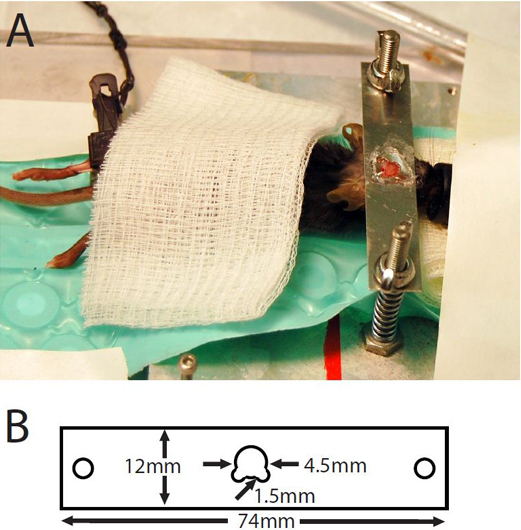
Figure 1. (A) Mouse with cranial window prepared for imaging. (B) Dimensions of head plate.
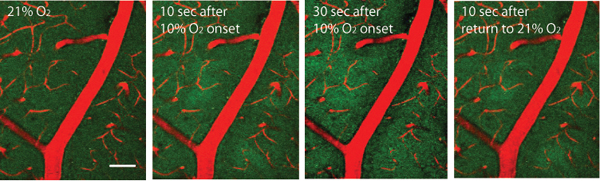
Figure 2. Endogenous NADH green fluorescence visualized with two-photon imaging through cranial window, approximately 50 μm below the pial surface. Blood vessels are visualized with Texas Red dextran. Oxygen in inhaled air was reduced to 10% for 3 min, and then restored for 21%. Scale bar: 50μm.
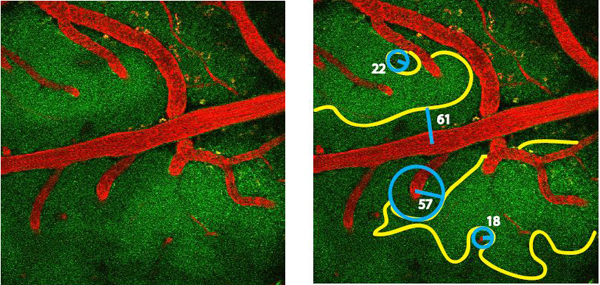
Figure 3. Geometry of NADH fluorescence boundaries. Yellow outline shows boundary of functional hypoxia; blue circles show projected oxygenated (Krogh) cylinders. Blue lines show cylinder radii; numbers show radii in micrometers.
