The following results illustrate the expected outcomes of the described protocol, and in the case of PB2, the observed outcomes.
Using Gene Composer, five full-length target amino acid sequences of the influenza virus polymerase subunit PB2 were designed (Figure 2). The PB2 sequences were back translated and subjected to many engineering steps 3, resulting in codon harmonized sequences optimized for expression in E. coli. From the iPCR products (Figure 3b), a total of thirty-four constructs were successfully cloned into a modified pET28 vector system 10 with an N-terminal 6x His-Smt fusion tag using PIPE cloning 3 as shown in Figure 3a. A summary of the cloning workflow is presented in Figure 4.
After successful cloning, micro-scale protein expression of each construct was tested in BL21(DE3) E. coli cells. Cells were grown in TB medium supplemented with Novagen Overnight Express 1 medium (according to the manufacturer’s protocol) for 48 hr at 20 °C in a shaking incubator set at 220 rpm. After growth, cells were harvested and tested for soluble protein expression using capillary electrophoreses with a Caliper LabChip 90. Fourteen of the thirty-four PB2 constructs led to soluble target protein and entered large-scale fermentation. Large-scale cultures of each construct were grown in TB medium supplemented with Novagen’s Overnight Express 1 medium according to the manufacturer’s protocol. After growth, cells were harvested via centrifugation and stored at -80 °C. Large-scale protein expression of each culture was confirmed via SDS-PAGE analysis (Figure 5) before proceeding with large-scale purification.
The Protein Maker was used to conduct parallel purification of the fourteen PB2 constructs. The clarified lysates of all fourteen constructs were run through a nickel-chelate column. After determining which fractions contained target protein by SDS-PAGE, the corresponding fractions were pooled for each sample and the concentration of each was determined by an A280 reading. Removal of the 6x His-Smt tag was conducted by the addition of ULP1 followed by overnight dialysis and a second nickel column. Confirmation of the His-Smt tag removal was conducted by SDS-PAGE (Figure 6), and each sample was concentrated with a 10 kDa Amicon Ultra centrifuge tube. After concentration using the Amicon Ultra centrifuge tubes, each sample was run over a sizing column to achieve crystallographic purity. A second concentration was conducted to increase the protein concentration to a level necessary for crystallization. All fourteen constructs were successfully purified and entered into crystallization trials.
Crystallization was initiated by thawing the previously frozen protein. Crystallization was performed in a climate controlled room at 16 °C with specially designed plates (Emerald Bio) for sitting drop vapor diffusion (Figure 7). Initial screening was conducted with four sparse-matrix screens; JCSG+, Pact, Wizard Full, and CryoFull (Emerald Bio), following an extended Newman strategy. 0.4 μl of protein solution was then mixed with 0.4 μl of crystallant (or reservoir solution) from the corresponding reservoir using 96-well Compact Jr crystallization plates (Emerald Bio). Of the fourteen purified samples nine of them yielded crystals suitable for diffraction studies (Figure 8). An in-house diffraction data set was collected on five of the nine constructs crystallized at Cu Kα wavelength using a Rigaku SuperBright FR-E+ rotating-anode X-ray generator equipped with Osmic VariMax HF optics and a Saturn 944+ CCD detector (Figure 9). Each data set was processed with XDS/XSCALE4 and scaled to a final resolution. Attempts to solve the structures by molecular replacement were carried out with Phaser 5 from the CCP4 suite 7. The final models were obtained after refinement in REFMAC 7 and manual rebuilding with Coot 11. The structures were assessed and corrected for geometry and fitness withMolProbity 9. A total of four structures of the PB2 subunit were determined (Figure 10) and deposited into the PDB. Figure 11 illustrates the overall outcome at each stage in the MTPP pipeline.
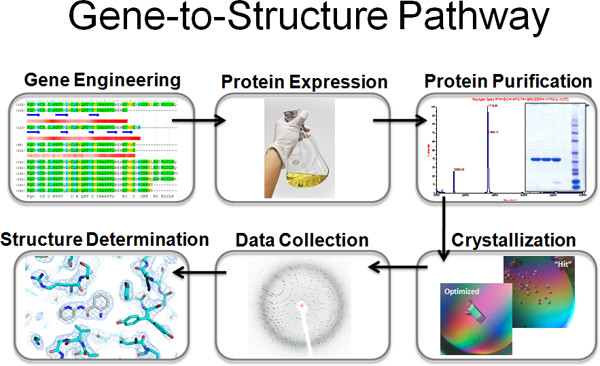
Figure 1. Overview of the SSGCID gene-to-structure pathway for Multi-target parallel processing at Emerald Bio.
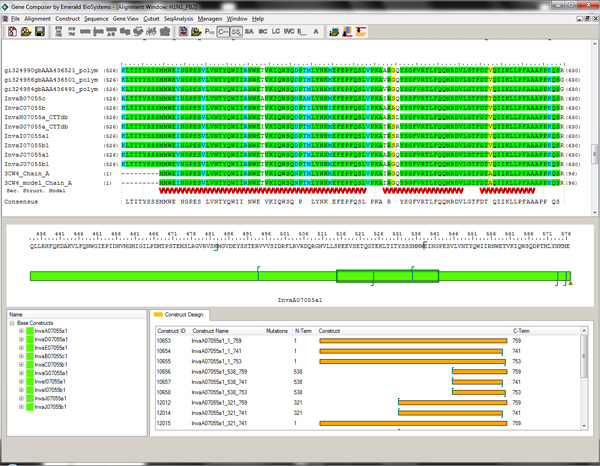
Figure 2. Alignment Viewer and Protein Construct Design Module in Gene Composer software. Amino-acid base construct of target is shown in green (middle window) and the structure guided truncations of alternative constructs are shown in gold (bottom window). An alignment of multiple Flu viral PB2 sequences is shown compared to the sequence and secondary structure elements of the C-terminal domain from PDBID 3CW4. Knowledge of the domain structure and secondary structure elements allows N-terminal truncations to be chosen within the Gene Composer Design Module by right-clicking on the desired amino acid residue. Click here to view larger figure.
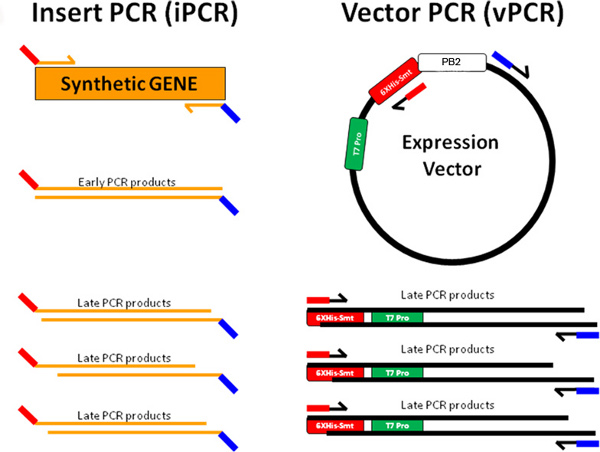
Figure 3a. PIPE cloning is illustrated wherein the synthetic gene insert (orange) is amplified by designed forward (red-orange lines) and reverse (orange-blue lines) primers to generate insert PCR material. The expression vector is amplified with reverse (red-black lines) and forward (blue-black lines) primers to generate vector PCR material. The terminal sequences iPCR products are complementary to the terminal sequences of vPCR products (red of iPCR complements red of vPCR and blue of iPCR complements blue of vPCR). This allows the iPCR and vPCR products to anneal to form plasmids that are replicated upon transformation into host BL21(DE3) chemically competent E. coli cells.
Figure 3b. Agarose gel analysis of iPCR products from the PB2 subunit. iPCR failures may be seen as faint or smeary bands, while successful iPCR products are represented by robust bands. iPCR product quality can generally be correlated with cloning success. Molecular weight markers are in kiloDaltons. Figure is reproduced from Raymond et al., 2011 12.
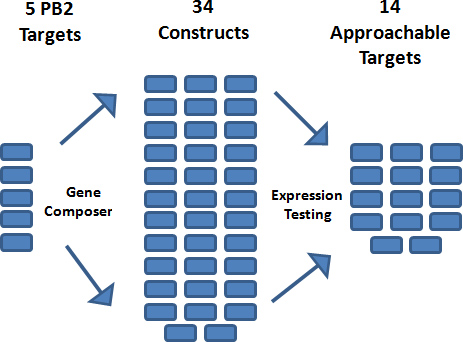
Figure 4. Gene engineering steps of target PB2 proteins were performed using Gene Composer software. After the engineered nucleic acid sequence was established for each target, 6-7 alternative protein constructs were designed for each. Multi-target parallel processing in the initial steps of gene design and cloning resulted in 34 constructs, 14 of which were viable targets that produced soluble proteins in E. coli.
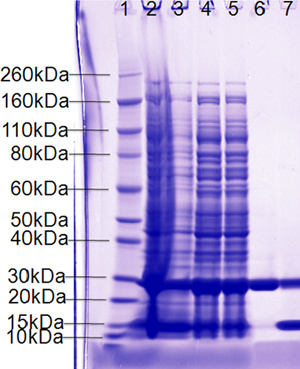
Figure 5. Representative SDS-PAGE analysis of large scale fermentation showing robust protein expression (expected size of 25.76 kDa), roughly 50% soluble (lane 4) and about 50% cleavage of 6x His-Smt tag from eluted protein (lane 7).
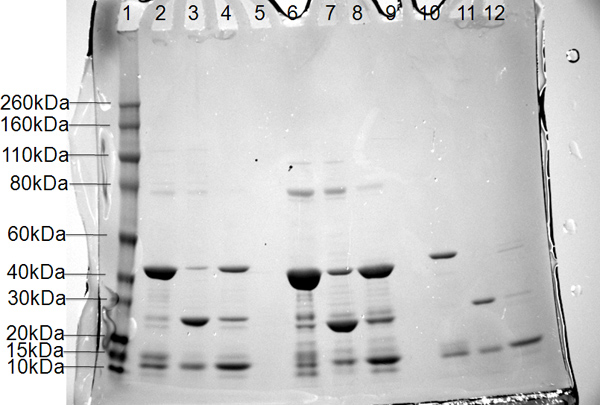
Figure 6. SDS-PAGE results for three constructs of the polymerase PB2 subunit. Lane 1, molecular-weight markers (labeled on the left in kDa); lanes 2, 6, and 10, pooled protein from Nickel 1 column; lanes 3, 7, and 11, flow-through of cleaved protein in buffer A from Nickel 2; lanes 4, 8, and 12, removal of 6x His-Smt tag in buffer B from Nickel 2.
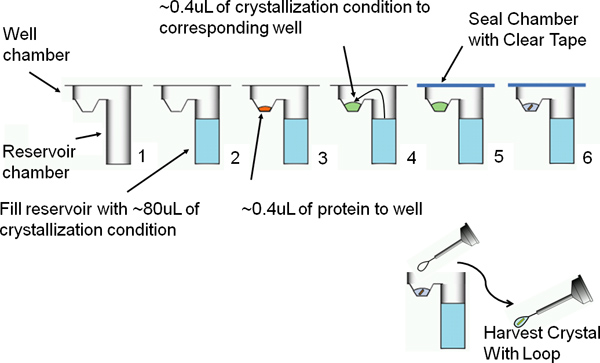
Figure 7. A schematic of vapor diffusion by the sitting drop method. The sitting drop method for protein crystallization falls under the category of vapor diffusion. This method entails a purified sample of protein and precipitant to equilibrate with a larger reservoir containing similar conditions in a higher concentration. As water vaporizes from the protein sample and transfers to the reservoir, the precipitant concentration increases to an optimal level for protein crystallization.
Figure 8. Protein crystal of polymerase PB2 subunit from a strain of the influenza virus.

Figure 9. X-ray diffraction image of the polymerase PB2 subunit from a strain of the influenza virus.
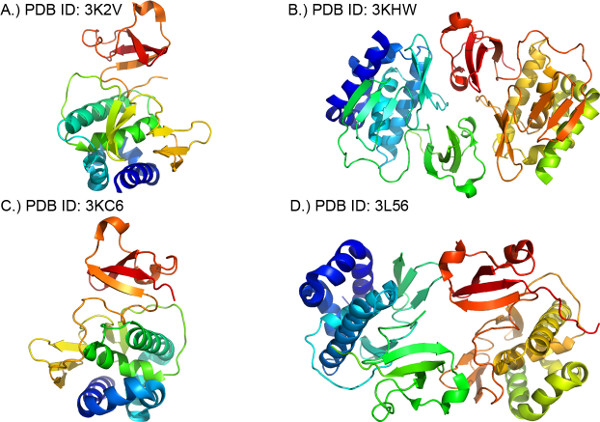
Figure 10. Ribbon diagrams of the molecules in the crystallographic asymmetric unit of 4 PB2 structures. Secondary structures colored in rainbow pattern with corresponding PDB codes. (a) 3K2V (A/Yokohama/2017/2003/H3N2) (b) 3KHW (A/Mexico/InDRE4487/2009/H1N1) (c) 3KC6 (A/Vietnam/1203/2004/H5N1) (d) 3L56 (A/Vietnam/1203/2004/H5N1).
Figure 11. Outcome analysis for influenza PB2 targets by the methods described. The structure determination pipeline is illustrated in five steps: Cloning, solubility, purification, crystallization and structure determination.
