The use of tissue specific probes can provide outstanding information in regards to the state of development for specific organs. In the following examples, the stage of the embryo is based on the Nieuwkoop and Faber staging table 11. If one uses probes form genes expressed after differentiation, cardiac troponin I at stage 28-30, for example (Figure 1C), the presence or size of a differentiated organ can be assessed at any stage post differentiation. Years ago, embryologists were able to do such analysis based on remarkable knowledge of the histology of the early embryo 12,13 but that expertise has been largely lost to the current generation of embryologists. Although, the loss of this expertise can be considered regrettable, the reliability and ease of use of in situ hybridization techniques makes identification of specific tissues available to any researcher. Tissues that are relatively inconspicuous, the hatching gland at embryonic stage 25 for example (Figure 1A), can be vividly marked using in situs without the need for specific antibodies or histological techniques (Figure 1C). Also, the use of whole mount in situs allows one to view the entire organ in the context of the whole embryo rather than relying on inference from histological sections (Figure 2). Even tissues that are deep within the embryo including the optic stalk (Figure 4) can be viewed easily and the use of embryo clearing can provide sharp delineation of the organ boundaries.
Bleaching of embryos to remove endogenous pigment using peroxide solutions has largely eliminated the requirement for using albino embryos that lack pigment. Bleaching for different times can be useful. For example, if the stain is strong, a lighter bleaching that allows for some pigmentation to be seen can be useful because it allows for better staging and orientation of the embryo (Figure 2A). However, if the stain is an area where there are high levels of pigmentation, such as the kidney (Figure 2B), near complete elimination of the pigment by longer incubation in the bleaching solution gives a better result.
Embryos tend to take up particular positions when in solution. After neurulation, they tend to lay flat on their sides. This is fine for images of the flank (Figure 2B) but can be a problem with other areas. Use of an agarose base allows one to cut channels into the agarose that can be use to orient the embryos. For example, blood precursors are localized to the ventral side of the embryo (Figure 2A) and the full extent of the staining is hard to observe. Positioning of the embryo in a channel with the ventral side up, then allows for full viewing of that gene expression pattern (Figure 3A).
The use of double in situ hybridization can show the relationship between two gene expression patterns within a single organism (Figure 5) eliminating the necessity of comparing between different embryos that may have small differences in morphology. Perhaps most importantly, the use of in situ hybridization gives one the ability to clearly mark cells prior to clear histological differentiation based on the expression of genes that are expressed in early progenitors of a lineage, such as pax2 that is expressed in many tissues in the early embryo, prior to differentiation (Figure 4A). The ability to identify progenitors and tissues that are not clearly distinguished based on histology, such as myeloid cell precursors (Figure 1B), has allowed researchers to ask much more detailed questions about the state of an organ’s development and also to assess the results of experimental manipulation designed to cause ectopic differentiation of a tissue.
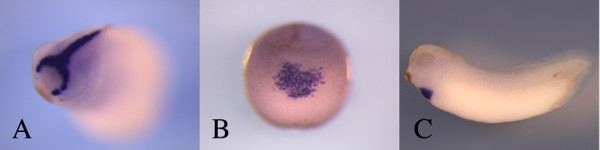
Figure 1: Examples of the whole mount in situ hybridization on Xenopus embryos. The blue staining from an in situ hybridization experiment can clearly delineate developing structures of the early Xenopus embryo. (A) An anterior view of a stage 26 embryo highlighting the hatching gland as demarcated by the expression of uvs.2 14. (B) A ventral view of an embryos showing the location of early myeloid cells at stage 20 using the expression of myeloperoxidase15 as a marker. (C) The early heart at stage 28 – 30 is visualized by the expression of cardiac troponin I 16. A clear advantage of using this method is that all of these gene expression patterns were visualized using different probes but the protocol used is identical in all cases. Please click here to view a larger version of this figure.
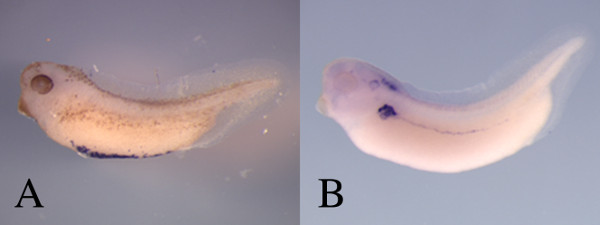
Figure 2: Different levels of bleaching can be used to visualize staining in the embryo. (A) This blue staining along the bottom of this embryo shows the expression of hemoglobin in the ventral blood islands at about stage 36. This is a region of the embryo that is only lightly pigmented and thus the embryo was not bleached for a long time as can be seen by the tan colored pigment in the eye and along the flank of the embryo. Being able to see the pigmentation allows for better visualization of the stage of the embryo. If the staining is in a region with greater natural pigmentation, such as the nervous system and the flank of the embryo, greater bleaching will help view the in situ as seen in B. (B) Here the expression of pax8 17 in the forming kidney, pronephric duct and hindbrain is best visualized after bleaching has removed almost all endogenous pigment. Please click here to view a larger version of this figure.
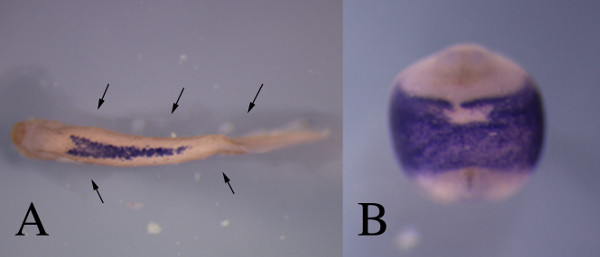
Figure 3: Manipulation of the agarose base can help with orientation of the embryos. Imaging specific regions of the embryo can be difficult because they tend to take up particular positions in the dish. (A) At tadpole stages the embryo will lie on its side. By cutting a fine channel in the agarose (black arrows) the embryo can be viewed from the ventral side, here showing hemoglobin expression at stage 36, with enough stability to capture a good image. (B) This ventral view of the hand1 expression at stage 20 outlines the lateral plate mesoderm 6. The embryo is placed in a small hole that stabilized its position with the ventral side up. Please click here to view a larger version of this figure.
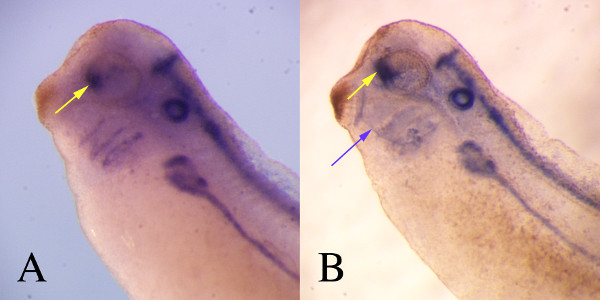
Figure 4: Internal organs can be viewed in both opaque, uncleared and in cleared embryos. In an uncleared embryo stained for pax2 at stage 34 (A), the optic stalk (yellow arrow) can be visualized relatively easily as can the staining down the neural tube. However, details are not sharp. By clearing the embryo (B) the boundaries of expression sites, including the optic stalk (yellow arrow) are sharper. The extent of clearing is shown by the ability to see both eyes in this cleared embryo. Also note that some staining has accumulated in the internal cavities (purple arrow), a common problem when viewing cleared embryos.
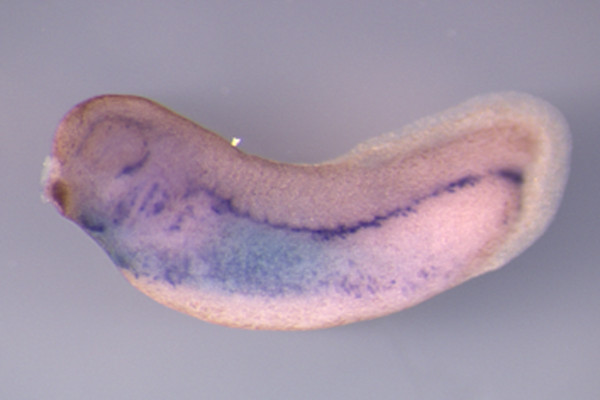
Figure 5: A representative double in situ hybridization on a Xenopus embryo. Expression of the lateral plate marker, hand1, is visualized by the light blue staining at stage 26. Expression of etv2 within the developing vasculature is visualized by dark blue purple stain. Expression of hand1 is usually very intense and thus it was utilized for the weaker fluorescein-labelled probe and BCIP combination. The utilized the digoxigenin-labelled probe and BM-purple combination for the weaker etv2 expression.
| Name | Final Concentration | Amount/100 ml |
| Mempfa | 1 mM MgSO4 | 100 µl of 1 M MgSO4 |
| (store at 4°C) | 2 mM EGTA, pH 8.0 | 10 ml of 20 mM EGTA, pH 8.0 |
| 0.1 M MOPS, pH 7.5 | 10 ml of 1 M MOPS, pH 7.5 | |
| 4% Paraformaldehyde, pH 7.5 | 50 ml of 8% Paraformaldehyde, pH 7.5 | |
| TTw | 50 mM Tris, pH 7.4 | 5 ml of 1 M Tris, pH 7.4 |
| 200 mM NaCl | 4 ml of 5 M NaCl | |
| 0.1% Tween 20 | 100 µl of Tween 20 | |
| 100X Denhardt's Solution | 2 % BSA | 2 g BSA |
| (filter through 0.2 µm filter, | 2% PVP-40 | 2 g PVP-40 |
| store at -20°C) | 2% Ficoll 400 | 2 g Ficoll 400 |
| 20X SSC | 3 M NaCl | 17.5 g NaCl |
| 300 mM Trisodium Citrate | 8.8 g Trisodium Citrate | |
| RNA Hybridization Buffer | 50% formamide | 50 ml of 100% formamide |
| (store at 4°C) | 5x SSC | 25 ml of 20x SSC |
| 1 mg/ml yeast RNA | 4 ml of 1mg/ml yeast RNA dissolved in 50% formamide | |
| 1X Denhardt's Solution | 1 ml of 100x Denhardt's Solution | |
| 0.1% Tween 20 | 100 µl of Tween 20 | |
| 5 mM EDTA, pH 8.0 | 1 ml of 0.5 M EDTA, pH 8.0 | |
| MAB | 100 mM Maleic Acid | 1.16 g of Maleic Acid |
| (pH 7.5, store at 4°C) | 150 mM NaCl | 0.88 g of NaCl |
| MAB+HTSS+BR | 100 mM Maleic Acid | 96 ml of MAB |
| 4% Heat Treated Sheep Serum | 4 ml of Heat Treated Sheep Serum (heat treated at 55°C for 30 min and aliquoted) | |
| 2% Blocking Reagent | 2 g of Roche Blocking Reagent | |
| MAB+HTSS+BR+anti-Dig antibody | 100 mM Maleic Acid | 96 ml of MAB |
| 4% Heat Treated Sheep Serum | 4 ml of Heat Treated Sheep Serum | |
| 2% Blocking Reagent | 2 g of Roche Blocking Reagent | |
| 1:10,000 Anti-Dig-AP, Fab Fragments Antibody | 1.5 µl of Anti-Digoxygenin-AP, Fab Fragments Antibody | |
| Alkaline Phosphatase (AP) Buffer | 100 mM Tris, pH 9.5 | 10 ml of 1 M Tris, pH 9.5 |
| 50 mM MgCl2 | 5 ml of 1 M MgCl2 | |
| 100 mM Nacl | 2.5 ml of 4 M NaCl | |
| 0.1% Tween 20 | 100 µl of Tween 20 | |
| Bleaching Solution | 0.3% H2O2 | 3.34 ml of 30% H2O2 |
| 5% Formamide | 5 ml of 100% Formamide | |
| 0.5% SSC | 2.5 ml of 20x SSC | |
| Clearing Solution | 1/3 Benzyl Alcohol | 33 ml |
| 2/3 Benzyl Benzoate | 67 ml |
Table 1: Solution Recipes
| Name | Amount |
| Dig-NTP Mix | 5 µl of 20 mM CTP |
| (40 µl reaction) | 5 µl of 20 mM GTP |
| 5 µl of 20 mM ATP | |
| 3.25 µl of 20mM UTP | |
| 3.5 µl of 10mM Dig-11-UTP | |
| 18.25 µl distilled, autoclaved water | |
| Probe Synthesis | x µl of template DNA (dependent on concentration) |
| (20 µl reaction) | x µl of distilled, autoclaved water |
| 4 µl of Dig-NTP mix | |
| 0.5 µl of RNAse inhibitor | |
| 2 µl of 10X RNA polymerase buffer | |
| 2 µl of RNA polymerase |
Table 2: Probe Synthesis Recipes
| Name of the Reagent | Approximate Volume | Duration | Temperature |
| 100% Methanol | 2 ml | 5 min, rocking | Room Temperature |
| 75% Methanol | 2 ml | 5 min, rocking | Room Temperature |
| 50% Methanol | 2 ml | 5 min, rocking | Room Temperature |
| 25% Methanol | 2 ml | 5 min, rocking | Room Temperature |
| TTw | 2 ml | 10 min, rocking | Room Temperature |
| TTw | 2 ml | 10 min, rocking | Room Temperature |
| TTw | 2 ml | 10 min, rocking | Room Temperature |
| Pre-warm RNA Hybridization Buffer and Probe to 65°C | |||
| TTw | 4 ml | 5 min, rocking | Room Temperature |
| TTw | 4 ml | 5 min, rocking | Room Temperature |
| RNA Hybridization Buffer | 2 ml | 10 min, rocking | Room Temperature |
| Pre-warmed RNA Hybridization Buffer | 2 ml | 1 hr, rocking | 65°C |
| Probe Solution | 1 ml | Overnight, rocking | 65°C |
Table 3: Steps for First Day of In Situ Hybridization Protocol (about 3 hr total)
| Name of the Reagent | Approximate Volume | Duration | Temperature |
| Pre-warm RNA Hybridization Buffer and 02.x SSC to 65°C and 2x SSC to 37°C | |||
| Return probe solution to tube for repeat use | |||
| RNA Hybridization Buffer | 2 ml | 10 min, rocking | 65°C |
| 2X SSC | 2 ml | 20 min, rocking | 37°C |
| 2X SSC | 2 ml | 20 min, rocking | 37°C |
| 0.2x SSC | 4 ml | 1 hr, rocking | 65°C |
| 0.2x SSC | 4 ml | 1 hr, rocking | 65°C |
| MAB+HTSS+BR | 1.5 ml | 30 min, rocking | Room Temperature |
| MAB+HTSS+BR+anti-DIG antibody | 1.5 ml | Overnight, rocking | 4°C |
Table 4: Steps for Second Day of In Situ Hybridization Protocol (about 4 hr total)
| Name of the Reagent | Approximate Volume | Duration | Temperature |
| MAB | 4 ml | 30 min, rocking | Room Temperature |
| MAB | 4 ml | 30 min, rocking | Room Temperature |
| MAB | 4 ml | 30 min, rocking | Room Temperature |
| MAB | 4 ml | 30 min, rocking | Room Temperature |
| MAB | 4 ml | 30 min, rocking | Room Temperature |
| MAB | 4 ml | 30 min, rocking | Room Temperature |
| MAB | 4 ml | 30 min, rocking | Room Temperature |
| MAB | 4 ml | 30 min, rocking | Room Temperature |
| MAB | 4 ml | 30 min, rocking | Room Temperature |
| MAB | 4 ml | 30 min, rocking | Room Temperature |
| MAB | 4 ml | 30 min, rocking | Room Temperature |
| MAB | 4 ml | 30 min, rocking | Room Temperature |
| BM Purple AP Substrate | 500-750 µl | Overnight, rocking (see text) | Room Temperature/37 °C (see text) |
Table 5: Steps for Third Day of In Situ Hybridization Protocol (about 7 hr)
| Name of the Reagent | Approximate Volume | Duration | Temperature |
| 25% Methanol | 2 ml | 5 min, rocking | Room Temperature |
| 50% Methanol | 2 ml | 5 min, rocking | Room Temperature |
| 75% Methanol | 2 ml | 5 min, rocking | Room Temperature |
| 100% Methanol (cold) | 2 ml | 20 min, rocking | Room Temperature |
| 100% Methanol (cold) | 2 ml | Varies, no rocking | Room Temperature |
| 75% Methanol | 2 ml | 5 min, rocking | Room Temperature |
| 50% Methanol | 2 ml | 5 min, rocking | Room Temperature |
| 25% Methanol | 2 ml | 5 min, rocking | Room Temperature |
| Mempfa | 2 ml | 30 min, rocking | Room Temperature |
| 25% Methanol | 2 ml | 5 min, rocking | Room Temperature |
| 25% Methanol | 2 ml | 5 min, rocking | Room Temperature |
| 25% Methanol | 2 ml | 5 min, rocking | Room Temperature |
| Bleaching Solution | 4 ml | 40 min-3 hr, rocking | Room Temperature/37 °C (see text) |
| Storing Embryos | |||
| 25% Methanol | 2 ml | 5 min, rocking | Room Temperature |
| 50% Methanol | 2 ml | 5 min, rocking | Room Temperature |
| 75% Methanol | 2 ml | 5 min, rocking | Room Temperature |
| 100% Methanol | 4 ml | Store at -20 °C | |
| Imaging Embryos | |||
| 1x PBS | 2 ml | 5 min, rocking | Room Temperature |
| 1x PBS | 2 ml | 5 min, rocking | Room Temperature |
| 1x PBS | 2 ml | 5 min, rocking | Room Temperature |
Table 6: Steps for Stopping In Situ Hybridization, Bleaching and Storage of Embryos
