Synthesis and Purification of Iodoaziridines Involving Quantitative Selection of the Optimal Stationary Phase for Chromatography
Summary
A protocol for the diastereoselective one-pot preparation of cis–N-Ts-iodoaziridines is described. The generation of diiodomethyllithium, addition to N-Ts aldimines and cyclization of the amino gem-diiodide intermediate to iodoaziridines is demonstrated. Also included is a protocol to rapidly and quantitatively assess the most appropriate stationary phase for purification by chromatography.
Abstract
The highly diastereoselective preparation of cis–N-Ts-iodoaziridines through reaction of diiodomethyllithium with N-Ts aldimines is described. Diiodomethyllithium is prepared by the deprotonation of diiodomethane with LiHMDS, in a THF/diethyl ether mixture, at -78 °C in the dark. These conditions are essential for the stability of the LiCHI2 reagent generated. The subsequent dropwise addition of N-Ts aldimines to the preformed diiodomethyllithium solution affords an amino-diiodide intermediate, which is not isolated. Rapid warming of the reaction mixture to 0 °C promotes cyclization to afford iodoaziridines with exclusive cis-diastereoselectivity. The addition and cyclization stages of the reaction are mediated in one reaction flask by careful temperature control.
Due to the sensitivity of the iodoaziridines to purification, assessment of suitable methods of purification is required. A protocol to assess the stability of sensitive compounds to stationary phases for column chromatography is described. This method is suitable to apply to new iodoaziridines, or other potentially sensitive novel compounds. Consequently this method may find application in range of synthetic projects. The procedure involves firstly the assessment of the reaction yield, prior to purification, by 1H NMR spectroscopy with comparison to an internal standard. Portions of impure product mixture are then exposed to slurries of various stationary phases appropriate for chromatography, in a solvent system suitable as the eluent in flash chromatography. After stirring for 30 min to mimic chromatography, followed by filtering, the samples are analyzed by 1H NMR spectroscopy. Calculated yields for each stationary phase are then compared to that initially obtained from the crude reaction mixture. The results obtained provide a quantitative assessment of the stability of the compound to the different stationary phases; hence the optimal can be selected. The choice of basic alumina, modified to activity IV, as a suitable stationary phase has allowed isolation of certain iodoaziridines in excellent yield and purity.
Introduction
The aim of this method is to prepare iodoaziridines that offer potential for further functionalization to aziridine derivatives. The method incorporates a protocol for the quantitative selection of the optimal stationary phase for chromatography.
Aziridines, as three-membered rings, posses inherent ring strain that makes them important building blocks in organic chemistry1. They display a vast array of reactivity often involving aziridine ring opening2,3, particularly as intermediates in the synthesis of functionalized amines4,5, or the formation of other nitrogen containing heterocycles6,7. The synthesis of a range of aziridine derivatives by functionalization of a precursor containing an intact aziridine ring has emerged as a viable strategy8. Functional group–metal exchange, to generate an aziridinyl anion, and reaction with electrophiles has been shown to be effective9,10,11, and recently regio- and stereoselective deprotonation of N-protected aziridines has also been achieved12-15. Very recently, palladium catalyzed cross-coupling methods to form aryl aziridines from functionalized aziridine precursors has been developed by Vedejs16,17, and ourselves18.
The chemistry of heteroatom substituted aziridines opens up fascinating questions of reactivity and stability19. We have been interested in the preparation of iodoaziridines as a novel functional group that offers the potential to provide precursors to a wide range of derivatives with complementary reactivity to existing aziridine functionalization reactions. In 2012 we reported the first preparation of aryl N-Boc-iodoaziridines20, and very recently reported the preparation of aryl and alkyl substituted N-Ts-iodoaziridines21.
The method to access iodoaziridines uses diiodomethyllithium, a reagent which has recently also been employed in the preparation of diiodoalkanes22,23, diiodomethylsilanes22,24, and vinyl iodides25-27. The carbenoid-like nature of this reagent requires preparation and use at low temperatures22,28. The techniques and conditions used for the generation of diiodomethyllithium in the preparation of iodoaziridines are described below.
While silica has emerged as the material of choice for chromatography29, it proved to be unsuitable for the purification of the N-Ts-iodoaziridines. Silica gel is generally the first and only solid phase material employed in flash chromatography in organic chemistry due to the availability and effective separations. However, the acidic nature of silica gel can cause the decomposition of sensitive substrates during purification, preventing isolation of the desired material. While other stationary phases or modified silica gels are available for chromatography30, there was no way to assess compatibility of the target molecule to these different materials. Due to the sensitive nature of the iodoaziridines, we established a protocol to assess the stability of a compound to an array of stationary phases21, which is demonstrated here. This has potential for application in the synthesis of a wide range of compounds with sensitive functional groups. The following protocol provides efficient access to N-Ts iodoaziridines, allowing the diastereoselective synthesis of both alkyl and aromatic cis-iodoaziridines in high yield.
Protocol
1. Preparation of Iodoaziridines with Diiodomethyllithium
- Flame dry a 100 ml round bottom flask containing a stirrer bar and fitted with a septum, under a stream of argon, then allow to cool to room temperature under an argon atmosphere. NOTE: Glassware dried in an oven overnight (125 °C) and cooled to room temperature in an analogous fashion is also appropriate.
- To the flask, add 5.7 ml anhydrous THF and 2.7 ml anhydrous Et2O via syringe, and freshly distilled hexamethyldisilazane (1.50 mmol, 315 µl) via a microsyringe.
- Stir the resulting solution and cool to -78 °C in a dry ice/acetone bath in a suitably sized dewar to allow the flask to be well submerged. Cover the dewar with aluminum foil, to minimize exposure of the reaction vessel to light.
- Add nBuLi (1.50 mmol, 0.60 ml, 2.5 M in hexanes) dropwise via syringe over 2-3 min to the solution at -78 °C. Allow the mixture to stir at -78 °C for a further 30 min to form a 0.17 M solution of LiHMDS. CAUTION: nBuLi solution is flammable, corrosive to the skin and pyrophoric. Excess reagent in the syringe should be quenched accordingly.
- After 30 min, add 1 ml anhydrous THF to a flame-dried 10 ml round bottom flask via syringe, followed by diiodomethane (1.70 mmol, 135 µl) via a microsyringe and ensure they are well mixed.
- Add the diiodomethane solution dropwise over 2 min to the solution of lithium hexamethyldisilazane at -78 °C. Leave this solution for 20 min at -78 °C.
- During this time, weigh out N-[(E)-4-methylphenylmethylidene]-4-methylbenzenesulfonamide (137 mg, 0.50 mmol) into another flame-dried 10 ml round bottom flask and dissolve in 2.0 ml anhydrous THF.
- After the 20 min deprotonation time, add the imine solution dropwise to the diiodomethyllithium solution over 5 min at -78 °C.
- Immediately after the dropwise addition is complete, lift the reaction vessel out of the dry ice bath, and transfer to an ice/water bath at 0 °C. Re-cover with aluminum foil and leave for 15 min at 0 °C. NOTE: The solution should be orange in color.
- After 15 min at 0 °C, quench the reaction by the addition of 30 ml saturated aqueous sodium bicarbonate solution. Transfer the mixture to a separating funnel and add 30 ml CH2Cl2. Shake the mixture and remove the lower CH2Cl2 layer. Repeat this extraction procedure two further times, and combine the CH2Cl2 layers.
- Add sodium sulfate to the organic layers to remove any water present in the solution, then filter off the sodium sulfate and collect the filtrate in a 250 ml round bottom flask.
- Remove the solvent under reduced pressure on a rotary evaporator to afford an impure sample of the desired iodoaziridine product.
2. Assessment of Product Stability to Stationary Phases for Chromatography
- Dissolve the crude aziridine sample in CH2Cl2 (16 ml) and add 1,3,5-trimethoxybenzene (28.0 mg, 0.167 mmol) as an internal standard, ensuring this is fully dissolved. Take an aliquot (2 ml) from this mixture, remove the solvent under reduced pressure and analyze this sample by 1H NMR spectroscopy.
- Open the recorded 1H NMR spectrum using standard NMR processing software. In Mestrenova, right click the spectrum and chose “integration”, then “manual” to provide the integration tool. Click and drag to cover the width of the peaks at 6.08 ppm and at 4.87 ppm to integrate the signals of the internal standard and the aziridine CHI signal respectively. Right-click on the integral for the peak at 6.08 ppm, select “edit integral” and change the “normalized” value to 3.0. NOTE: Similar steps can be applied with other software packages.
- Use the updated value of integral for the aziridine CHI signal (4.87 ppm) to determine the yield of the iodoaziridine, here using (100/3) × (the integral of the CHI signal), which affords a calculated yield of 59%. NOTE: Given the known quantity of internal standard (0.167 mmol), and the product peak corresponding to 1 proton, the yield of iodoaziridine is calculated by the following equation:100 × (integral of product peak) × (moles of internal standard) / moles starting material.
- Prepare slurries of the following stationary phases (25 g): silica, silica + 1% NEt3 (triethylamine), neutral alumina, basic alumina (activity I), basic alumina (activity IV) and Florisil, each in 5% EtOAc/hexane (50 ml), in six separate 250 ml conical flasks containing stirrer bars. In another conical flask prepare a 5% EtOAc/hexane solution (50 ml), to be used as a control experiment. CAUTION: silica gel, alumina and other stationary phases employed are dangerous if inhaled, therefore should always be handled in an effective fume hood.
- Add 2 ml aliquots of the iodoaziridine/internal standard solution to each of the conical flasks at RT. Stir the slurry mixtures for 30 min. NOTE: this represents the duration the compound may be exposed to the stationary phase during a normal flash column chromatography procedure.
- Filter the slurry mixtures using a sintered funnel, and collect the filtrate in a 250 ml round bottom flask. Wash the residue on the sintered funnel with CH2Cl2 (2 × 30 ml). Repeat this filtering process for the remaining slurries. NOTE: It is appropriate to offset the commencement of each stationary phase to allow time for filtration and so maintain the same time for each of the stationary phase materials.
- Remove the solvent from the resulting samples under reduced pressure, and analyze by 1H NMR spectroscopy to calculate the amount of iodoaziridine recovered in each case, as described in Section 2.2.
- Compare the yields of iodoaziridine obtained from each stationary phase tested with that obtained in Section 2.1. NOTE: The sample giving the highest yield, ideally the same as in 2.1, indicates the optimal stationary phase for chromatography. In this example, basic alumina (activity IV) was deemed the best stationary phase for purification.
3. Deactivation of Basic Alumina and Purification of the Iodoaziridine
- Repeat Section 1 to generate the crude iodoaziridine mixture.
- To generate basic alumina (activity IV), add 100 g of basic alumina (activity I) to a 500 ml round bottom flask and then add 10 ml water to the flask and fit with a glass stopper.
- Shake the flask vigorously until no lumps are visible, indicating even spreading of water throughout the alumina. Allow the alumina to cool to RT. CAUTION: the adsorption of water is exothermic, so the flask may get hot and may result in a build up of pressure. Release any pressure build up frequently.
- Purify the crude iodoaziridine by column chromatography using the basic alumina (activity IV) as the stationary phase, eluting with hexane, grading to 5% EtOAc/hexane. NOTE: high concentrations of EtOAc should not be used with basic alumina. In these cases, diethyl ether can be used instead.
- Combine the product containing fractions and remove the solvent under reduced pressure to obtain the pure iodoaziridine.
Representative Results
The procedure described affords cis-(±)-2-iodo-3-(4-tolyl)-1-(4-tolylsulfonyl)aziridine as a single diastereoisomer and with excellent purity (Figure 1). Prior to purification, a yield of 59% of the iodoaziridine product was calculated by 1H NMR spectroscopy. However, this iodoaziridine was particularly challenging to purify and underwent significant decomposition on silica. Purification on basic alumina (activity IV) as determined by the stationary phase screen allowed the product to be isolated in 48% yield. The results from the stationary phase screen are illustrated in Figure 2. Following filtration, analysis of the 1H NMR spectra gives a series of yields for the different materials used, with respect to the internal standard. These yields are representative of the isolated yield that can be expected after column chromatography on that specific stationary phase. Basic alumina (activity IV) returns the highest yield (53%), which is closest to the yield calculated by 1H NMR. Therefore, basic alumina (activity IV) was chosen as the stationary phase for column chromatography for the purification of the N-Ts iodoaziridine. The isolated yields, following chromatography, are comparable with those predicted.
A wide selection of iodoaziridines can be accessed by this method in high yield (see Figure 3 for representative examples). Both alkyl and aromatic N-Ts imines are compatible with the reaction, including the sterically demanding tert-butyl and ortho-tolyl examples. The reaction is proposed to occur by deprotonation of diiodomethane by lithium hexamethyldisilazane at -78 °C, forming diiodomethyllithium (Figure 4). On addition of the N-Ts aldimine, nucleophilic addition of the diiodomethane anion to the imine at -78 °C affords the amino gem-diiodide intermediate. Subsequent warming to 0 °C induces a highly diastereoselective cyclization of the amino gem-diiodide intermediate, affording the cis–N-Ts-iodoaziridine exclusively. The cyclization occurs highly stereoselectively with the cis-iodoaziridine being favored over the trans-iodoaziridine due to subtle steric interactions in the cyclization transition state.
During reaction optimization, it was apparent that control of temperature and the timing of the different stages is essential to the outcome of the reaction (Figure 5). Quenching the reaction at -78 °C without warming results in the formation of the N-Ts iodoaziridine and the amino gem-diiodide. However, the products undergo degradation under the reaction conditions, which is avoided by warming and reducing the reaction times.
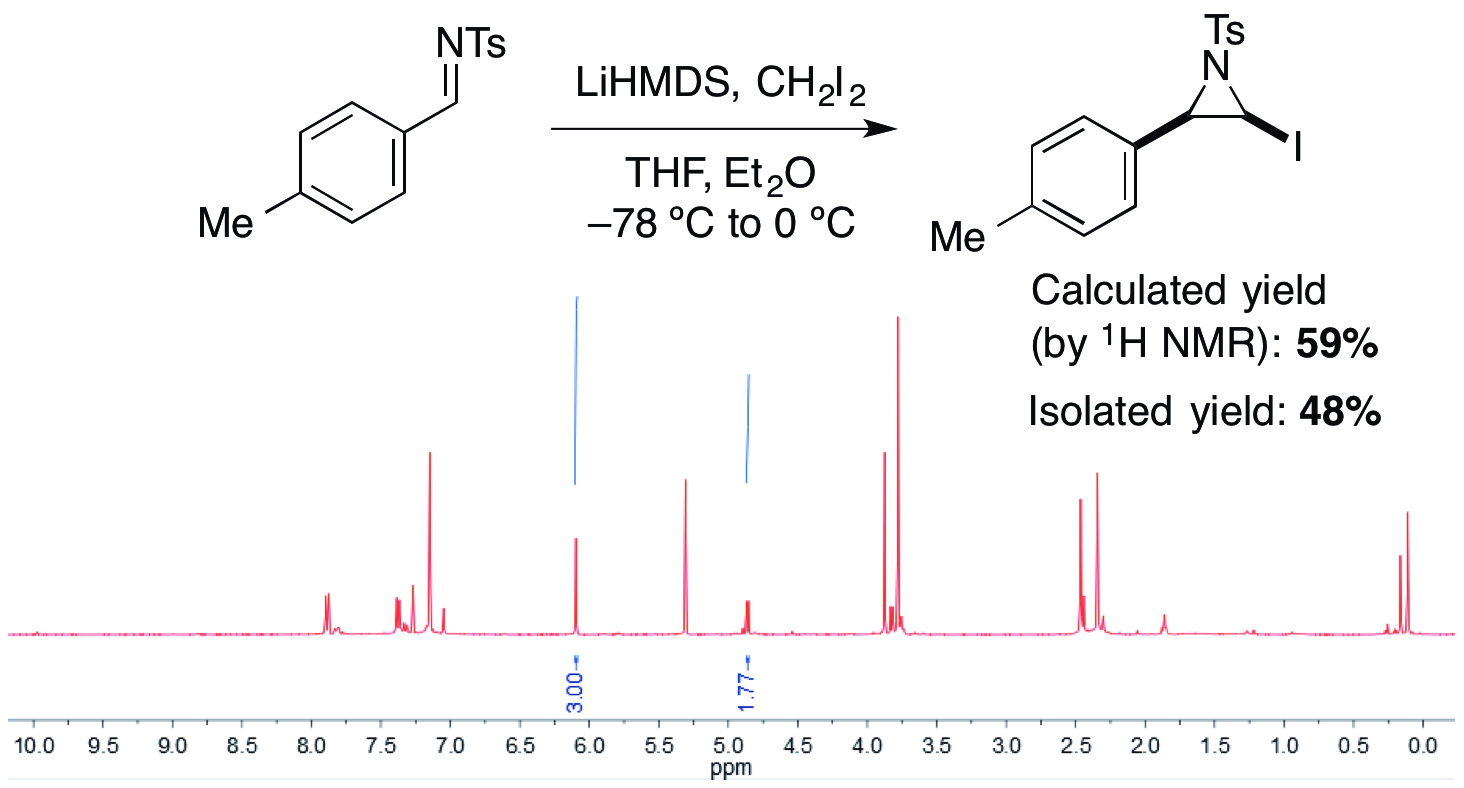
Figure 1. Formation of the para-tolyl iodoaziridine and the corresponding 1H NMR spectrum of the crude product mixture containing the iodoaziridine and 1,3,5-trimethoxybenzene.
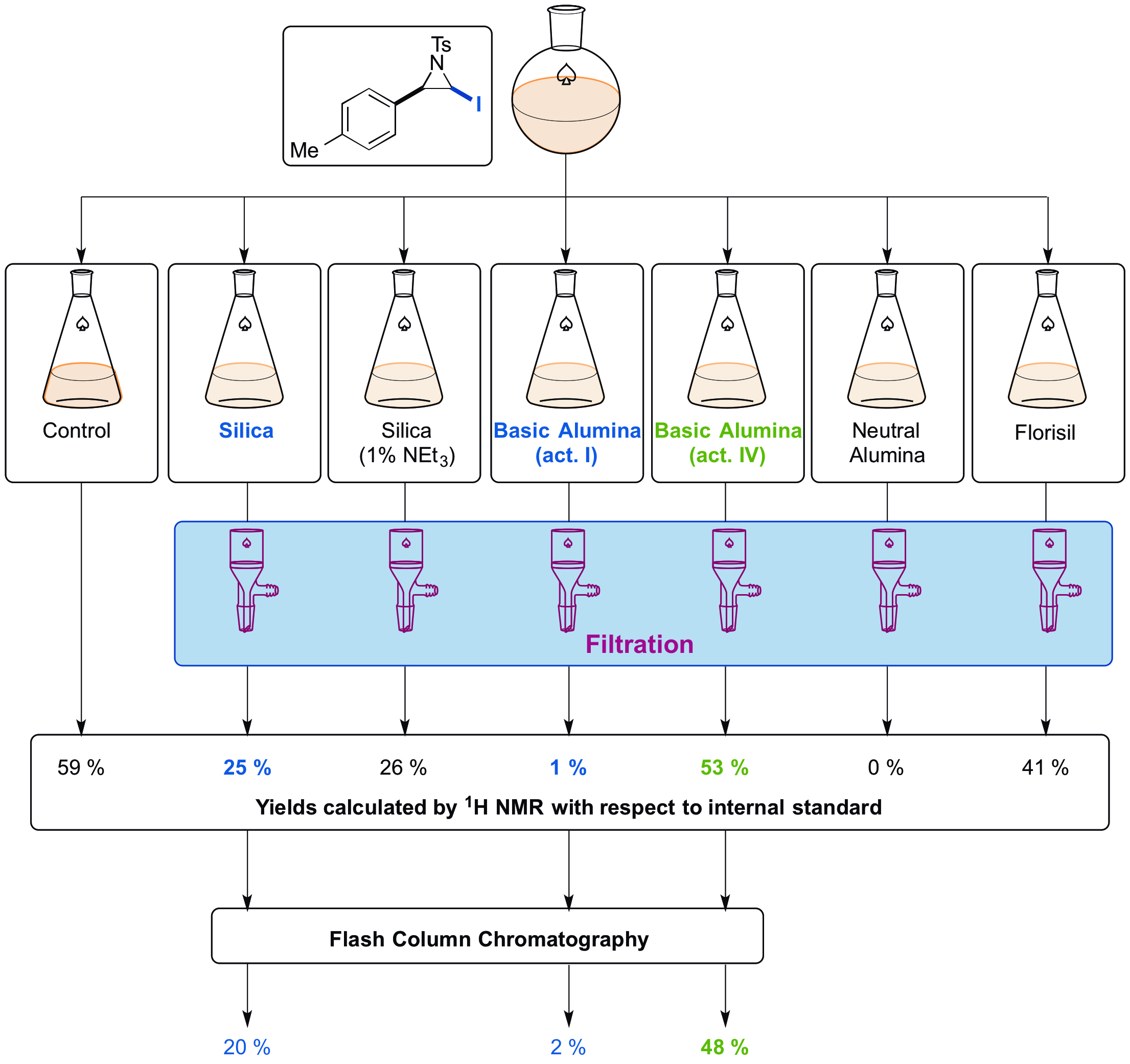
Figure 2. Process for 1H NMR stability study for the para-tolyl iodoaziridine with various stationary phases; the best recovery of iodoaziridine is observed using basic alumina (activity IV) (53%).
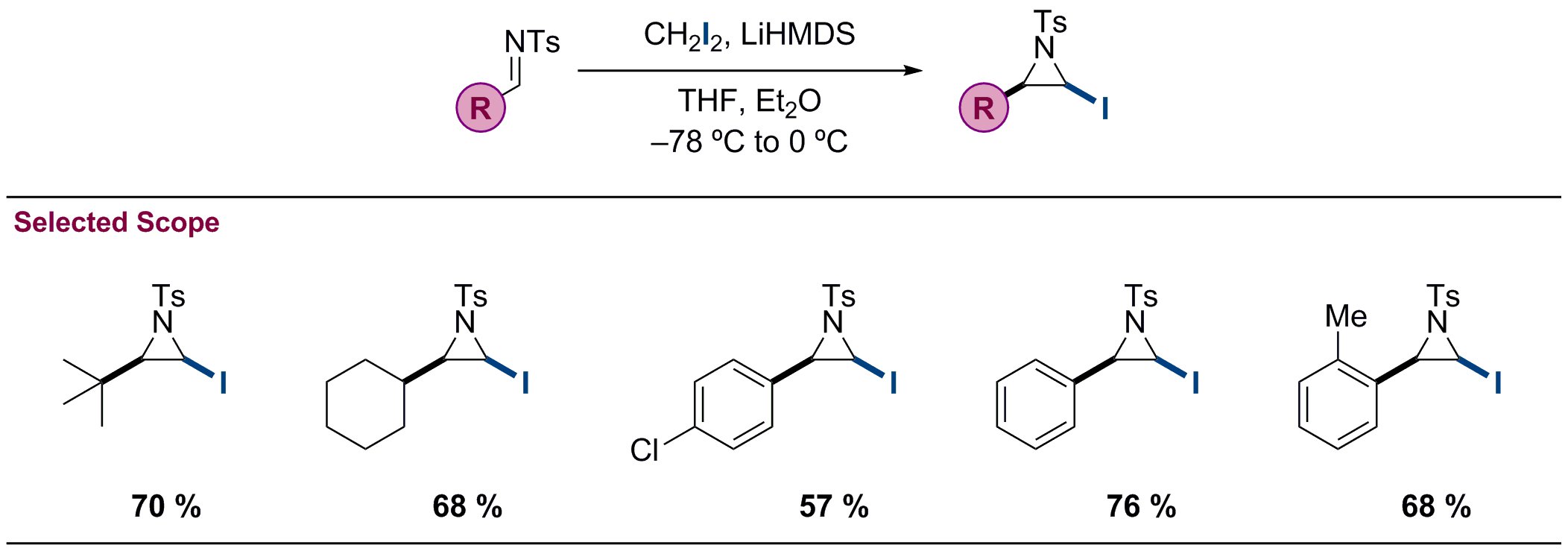
Figure 3. Selected scope of the iodoaziridination reaction.
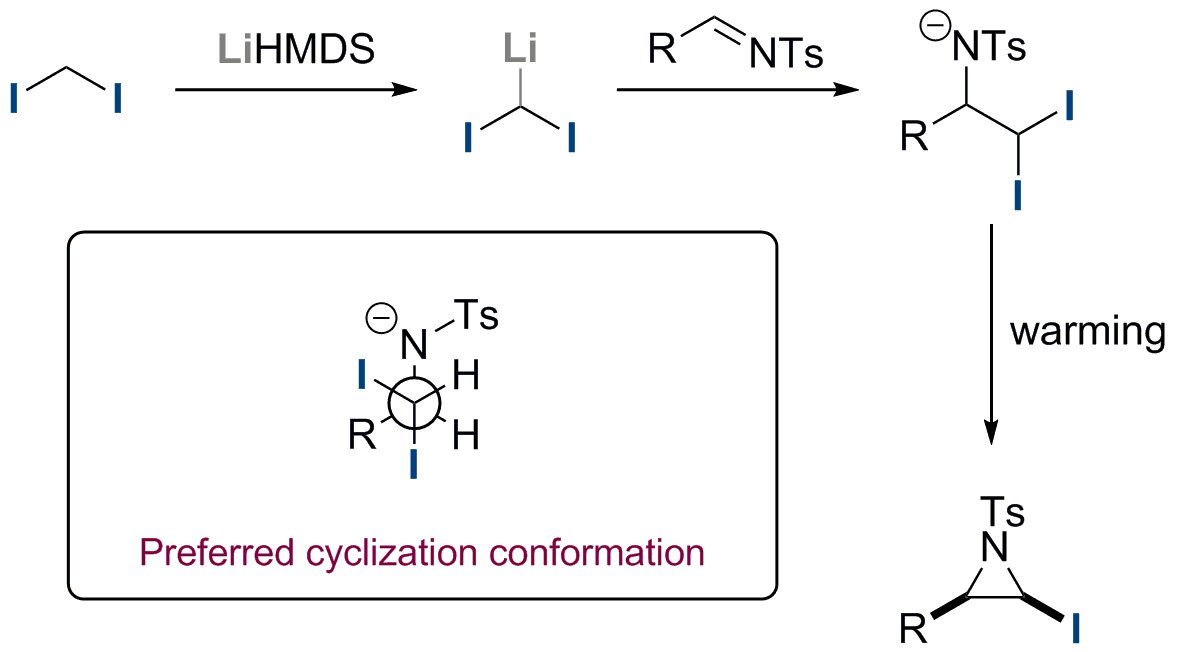
Figure 4. Proposed mechanism of reaction and rationale of diastereoselectivity.
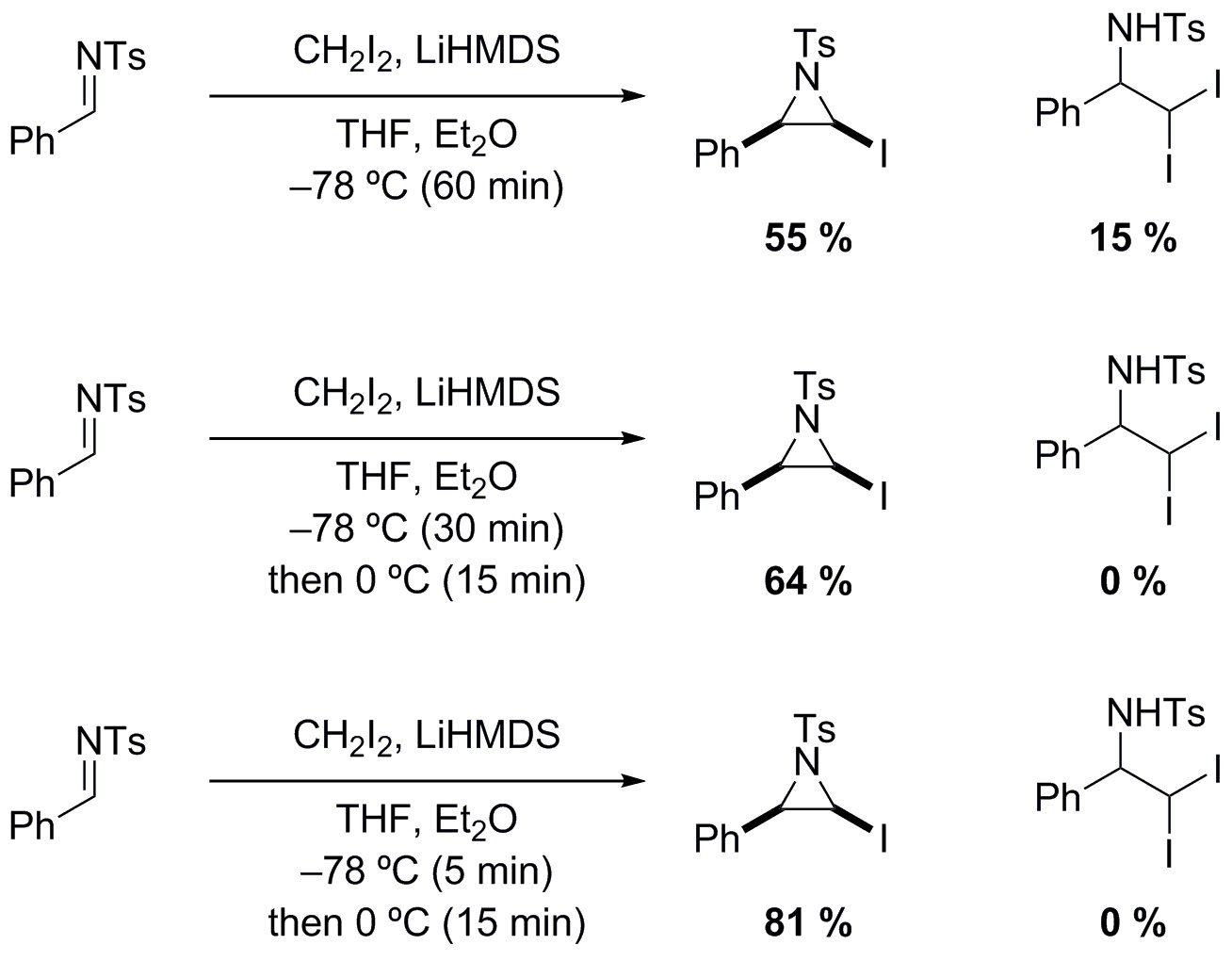
Figure 5. Ratio of iodoaziridine to amino gem-diiodide with varying reaction time and temperature.
Discussion
A procedure for the diastereoselective preparation of cis–N-Ts-iodoaziridines is described, along with a stability study protocol to quantitatively indicate the best stationary phase for purification of potentially unstable compounds by flash column chromatography. It is envisaged that access to iodoaziridines through this approach will enable methods to access to a wide range of aziridines to be developed, by derivatization of the intact ring.
An appropriate modification to the procedure for imines with an α-proton, is to use imine-toluene sulfinic acid adducts as starting materials in place of the imine due to the improved stability to storage and handling. From this starting material, an extra equivalent of both diiodomethane and LiHMDS should be employed to generate the imine in situ.
In preparation of the LiHMDS solution, the hexamethyldisilazane should be freshly distilled before use. Amine that has not been distilled can result in more of a minor aminal product being formed, through direct addition of the base into the aldimine. This aminal side product is also more prevalent when using commercial LiHMDS solutions, rather than a freshly prepared solution. Commercial nBuLi solutions must be regularly titrated to determine the concentration to control precisely the amount used in reaction. The diiodides and iodoaziridine products are light sensitive and so the reaction should be covered and exposure of the product to light should be minimized. Prolonged exposure to light leads to decomposition, so the isolated iodoaziridines should be stored at -20 °C in the dark.
The procedure described is limited to -branched imines with either the imine or imine-sulfinic acid adducts; only low yields are obtained for primary alkyl imines. This is due to the preferential direct addition of LiHMDS to the aldimine, over the desired addition of diiodomethyllithium, for less sterically hindered substrates.
To our knowledge there is not an available method to quantify the stability of a compound to stationary phases. This is particularly important for new compound classes or new small molecule functional groups. The protocol described here allows a rapid indication of the stability of the iodoaziridine to the various stationary phases, as well as providing a chance to identify decomposition products that could potentially be formed upon column chromatography. The protocol for quantitatively assessing the stability of iodoaziridines to stationary phases has potential for application in the purification of a wide range of compounds with sensitive functional groups, due to the general nature and ease of the setup.
There are a number of critical steps in the protocol. The dropwise addition of the imine/THF solution over 5 min is crucial to the yield of the product obtained. Faster addition times have shown to yield less of the desired iodoaziridine product. Purification on basic alumina (activity IV) is essential; use of silica results in decomposition products being observed. Basic alumina (activity IV) is not commercially available and must be prepared prior to use, as described in the protocol (3.2 and 3.3).
Divulgations
The authors have nothing to disclose.
Acknowledgements
For financial support we gratefully acknowledge the EPSRC (Career Acceleration Fellowship to J.A.B.; EP/J001538/1), the Ramsay Memorial Trust (Research Fellowship 2009-2011 to J.A.B.), and Imperial College London. Thank you to Prof. Alan Armstrong for generous support and advice.
Materials
| Hexamethyldisilazane | 999-97-3 | Alfa Aesar | Distill from KOH under argon prior to use. |
| n-Butyllithium | 109-72-8 | Sigma Aldrich | 2.5 M in hexanes, titrate prior to use. |
| Diiodomethane | 75-11-6 | Alfa Aesar | Contains copper as a stabilizer. |
| 1,3,5-Trimethoxybenzene | 621-23-8 | Sigma Aldrich | |
| Silica | 112945-52-5 | Merck | |
| Basic alumina | 1344-28-1 | Sigma Aldrich | |
| Neutral alumina | 1344-28-1 | Merck | |
| Florisil | 1343-88-0 | Sigma Aldrich | |
| THF | All anhydrous solvents were dried through activated alumina purification columns. | ||
| Et2O | |||
| CH2Cl2 | |||
| NMR spectrometer | Bruker AV 400 | n/a | |
| NMR processing software | MestReNova | 7.0.2-8636 | |
References
- Sweeney, J. B. Aziridines: epoxides’ ugly cousins. Chem. Soc. Rev. 31 (5), 247-258 (2002).
- Lu, P. Recent developments in regioselective ring opening of aziridines. Tetrahedron. 66 (14), 2549-2560 (2010).
- Wu, B., Parquette, J. R., RajanBabu, T. V. Regiodivergent ring opening of chiral aziridines. Science. 326 (5960), (2009).
- Liew, S. K., He, Z., St Denis, J. D., Yudin, A. K. Stereocontrolled synthesis of 1,2- and 1,3-diamine building blocks from aziridine aldehyde dimers. J. Org. Chem. , (2013).
- Stanković, S., et al. Regioselectivity in the ring opening of non-activated aziridines. Chem. Soc. Rev. 41 (2), 643-665 (2012).
- Cardoso, A. L., Pinho e Melo, T. M. V. D. Aziridines in formal [3+2] cycloadditions: synthesis of five-membered heterocycles. Eur. J. Org. Chem. 2012 (33), 6479-6501 (2012).
- Dauban, P., Malik, G. A masked 1,3-dipole revealed from aziridines. Angew. Chem., Int. Ed. 48 (48), 9026-9029 (2009).
- Florio, S., Luisi, R. Aziridinyl anions: generation, reactivity, and use in modern synthetic chemistry. Chem. Rev. 110 (9), 5128-5157 (2010).
- Vedejs, E., Moss, W. O. Lithiated aziridine reagents. J. Am. Chem. Soc. 115 (4), 1607-1608 (1993).
- Satoh, T., Fukuda, Y. A new synthesis of enantiomerically pure α- and β-amino acid derivatives using aziridinyl anions. Tetrahedron. 59 (49), 9803-9810 (2003).
- Satoh, T., Matsue, R., Fujii, T., Morikawa, S. Cross-coupling of nonstabilized aziridinylmagnesiums with alkylhalides catalyzed by Cu(I) iodide: a new synthesis of amines bearing a quaternary chiral center and an asymmetric synthesis of both enantiomers of the amines from one chiral starting material. Tetrahedron. 57 (18), 3891-3898 (2001).
- Hodgson, D. M., Humphreys, P. G., Hughes, S. P. Widening the usefulness of epoxides and aziridines in synthesis. Pure. Appl. Chem. 79 (2), 269-279 (2007).
- Musio, B., Clarkson, G. J., Shipman, M., Florio, S., Luisi, R. Synthesis of optically active arylaziridines by regio- and stereospecific lithiation of N-Bus-phenylaziridine. Org. Lett. 11 (2), 325-328 (2009).
- Beak, P., Wu, S., Yum, E. K., Jun, Y. M. Intramolecular cyclizations of -lithioamine synthetic equivalents: convenient syntheses of 3-, 5-, and 6-membered-ring heterocyclic nitrogen compounds and elaborations of 3-membered ring systems. J. Org. Chem. 59 (2), 276-277 (1994).
- Aggarwal, V. K., Alonso, E., Ferrara, M., Spey, S. E. Highly diastereoselective aziridination of imines with trimethylsilyldiazomethane. Subsequent silyl substitution with electrophiles, ring opening, and metalation of C-silylaziridines − a cornucopia of highly selective transformations. J. Org. Chem. 67 (7), 2335-2344 (2002).
- Nelson, J. M., Vedejs, E. Metalated aziridines for cross-coupling with aryl and alkenyl halides via palladium catalysis. Org. Lett. 12 (22), 5085-5087 (2010).
- Theddu, N., Vedejs, E. Stille coupling of an aziridinyl stannatrane. J. Org. Chem. 78 (10), 5061-5066 (2013).
- Hughes, M., Boultwood, T., Zeppetelli, G., Bull, J. A. Palladium-catalyzed cross-coupling of aziridinylmetal species, generated by sulfinyl−magnesium exchange, with aryl bromides: reaction optimization, scope, and kinetic investigations. J. Org. Chem. 78 (3), 844-854 (2013).
- Singh, G. S., D’hooghe, M., De Kimpe, N. Synthesis and reactivity of C-heteroatom-substituted aziridines. Chem. Rev. 107 (5), 2080-2135 (2007).
- Bull, J. A., Boultwood, T., Taylor, T. A. Highly cis-selective synthesis of iodo-aziridines using diiodomethyllithium and in situ generated N-Boc-imines. Chem. Commun. 48 (100), 12246-12248 (2012).
- Boultwood, T., Affron, D. P., Trowbridge, A. D., Bull, J. A. Synthesis of cis-C-iodo-N-tosyl-aziridines using diiodomethyllithium: reaction optimization, product scope and stability, and a protocol for selection of stationary phase for chromatography. J. Org. Chem. 78 (13), 6632-6647 (2013).
- Bull, J. A., Charette, A. B. Improved procedure for the synthesis of gem-diiodoalkanes by the alkylation of diiodomethane. scope and limitations. J. Org. Chem. 73 (20), 8097-8100 (2008).
- Bull, J. A., Charette, A. B. Intramolecular Simmons-Smith cyclopropanation. Studies into the reactivity of alkyl-substituted zinc carbenoids, effect of directing groups and synthesis of bicyclo[n.1.0]alkanes. J. Am. Chem. Soc. 132 (6), 1895-1902 (2010).
- Lim, D. S. W., Anderson, E. A. One-step preparation of functionalized (E)-vinylsilanes from aldehydes. Org. Lett. 13 (18), 4806-4809 (2011).
- Bull, J. A., Mousseau, J. J., Charette, A. B. Convenient one-pot synthesis of (E)-β-aryl vinyl halides from benzyl bromides and dihalomethanes. Org. Lett. 10 (23), 5485-5488 (2008).
- Bull, J. A., Mousseau, J. J., Charette, A. B. Preparation of (E)-(2-iodovinyl)benzene from benzyl bromide and diiodomethane. Org. Synth. 87, 170-177 (2010).
- Boxer, M. B., Yamamoto, H. Super silyl group for a sequential diastereoselective aldol-polyhalomethyllithium addition reaction. Org. Lett. 10 (3), 453-455 (2008).
- Seyferth, D., Lambert, R. L. Halomethyl-metal compounds: LXII. Preparation of diiodomethyl-metal compounds. J. Organomet. Chem. 54, 123-130 (1973).
- Still, W. C., Kahn, M., Mitra, A. Rapid chromatographic technique for preparative separations with moderate resolution. J. Org. Chem. 43 (14), 2923-2925 (1978).
- Armarego, W. L. F., Chai, L. L. C. . Purification of laboratory chemicals. , (2003).
