In Vivo 4-Dimensional Tracking of Hematopoietic Stem and Progenitor Cells in Adult Mouse Calvarial Bone Marrow
Summary
The nature of the interactions between hematopoietic stem and progenitor cells (HSPCs) and bone marrow niches is poorly understood. Custom hardware modifications and a multi-step acquisition protocol allow the use of two-photon and confocal microscopy to image ex vivo labeled HSPCs homed within bone marrow areas, tracking interactions and movement.
Abstract
Through a delicate balance between quiescence and proliferation, self renewal and production of differentiated progeny, hematopoietic stem cells (HSCs) maintain the turnover of all mature blood cell lineages. The coordination of the complex signals leading to specific HSC fates relies upon the interaction between HSCs and the intricate bone marrow microenvironment, which is still poorly understood[1-2].
We describe how by combining a newly developed specimen holder for stable animal positioning with multi-step confocal and two-photon in vivo imaging techniques, it is possible to obtain high-resolution 3D stacks containing HSPCs and their surrounding niches and to monitor them over time through multi-point time-lapse imaging. High definition imaging allows detecting ex vivo labeled hematopoietic stem and progenitor cells (HSPCs) residing within the bone marrow. Moreover, multi-point time-lapse 3D imaging, obtained with faster acquisition settings, provides accurate information about HSPC movement and the reciprocal interactions between HSPCs and stroma cells.
Tracking of HSPCs in relation to GFP positive osteoblastic cells is shown as an exemplary application of this method. This technique can be utilized to track any appropriately labeled hematopoietic or stromal cell of interest within the mouse calvarium bone marrow space.
Introduction
Despite stem cell niches having been recognized for decades3 and well characterized in many tissues such as the olfactory bulb, muscle fiber, hair follicle bulge or CNS subventricular zone4-7, the interactions between hematopoietic stem cells (HSCs) and bone marrow microenvironment (BM) are still poorly understood due to the difficulty of directly observing single cells through the bone as well as the extremely fluid nature of the tissue itself. It has been shown, through a number of functional studies based on ablating or overexpressing specific genes in either HSCs or the stromal cells of the microenvironment itself, that an intricate network of different cell types and regulatory signals is responsible for regulating the delicate balance between quiescence, proliferation, expansion and differentiation of HSCs1-2. The crosstalk between HSCs and their niches can be understood in depth only through direct observation. This is achievable thanks to the development of advanced imaging protocols, combining the available technology not only in terms of microscopy, but also of specimen holder solutions and of acquisition software.
Single cell resolution live imaging of transplanted hematopoietic stem and progenitor cells (HSPCs) in the BM compartment of the mouse calvaria bones showed that engrafting HSPCs localize in proximity of SDF-1 and VCAM-1-positive vessels8-9 and that more immature cells are found within 15 – 20 microns from GFP-positive osteoblastic cells (Col2.3-GFP transgenic mouse line, in which the osteoblast-restricted collagen 1α promoter drives GFP expression) while their progeny are more distal10. Similar observations were obtained from dissected and fractured femur bones11 and from thinned tibeal bones12. Imaging of calvarial bone marrow remains the least invasive approach to achieve direct visualization of HSPCs within their native microenvironment.
With any live specimen imaging there is an inherent need to keep the sample as still as possible to avoid any unnecessary artifacts due to sample movement. Breathing causes oscillatory movements of the mouse head, which need to be avoided when imaging calvarial bone marrow. Conventional stereotactic holders, used for example for brain imaging and electrophysiology, are not suitable for calvarium imaging because they obstruct the frontal bones. Starting from a mouse holder used to minimize distress during live imaging and electrophysiology studies13, a 2 component holder was developed, with a small piece fixed to the head of the mouse to create an imaging window connected to a larger arm ensuring steady hold with a heavy base plate which secures firmly into the microscope stage. Securing the head piece to the skull of the mouse allows free breathing while still immobilizing the head in place and adequately eliminating movements due to breathing. A 'lock-and-key' mechanism linking the head-piece to the holder body allows the size of the window to be minimized as well as increased accuracy of positioning, therefore simplifying the surgery, and the fit of the base plate into the stage allows for accurate alignment on the microscope. To accurately monitor motile cells, a multi-point time lapse protocol was designed to allow tracking of cells over time with 5 min intervals between time points. Here, DiD labeled HSCs are injected into col2.3GFP osteoblasitc reporter mice10,14 and imaged approximately 20 hr later. The mouse holder that was designed and the imaging settings required to perform rapid multi-point time-lapse imaging of the same areas over a period of multiple hr are described in detail.
Protocol
All procedures involving the use of animals are carried according to the local legislation. Our work is approved by the UK Home Office as well as the Imperial College ethical review panel. The allowed procedures are described in the project license documentation and follow guidelines that ensure the welfare of the animal at all times. Ensure to adhere to the legislation on animal experimentation of the country where the work is performed.
1. Labeling and Injection of HSPCs:
- Harvest cells as described by Lo Celso14-15.
- Prepare the cells at a concentration of 106 cells/ml in PBS. NOTE: Use a hemocytometer or the information provided by the cell sorter to count the cells; the number of cells to be labeled and injected varies depending on the cell type (stain for example 10,000 – 20,000 HSCs, 100,000 – 300,000 HPCs); if working with less than 105 cells, resuspend in 100 µl of PBS; it is important to have the cells in PBS without any serum, as serum inhibits the staining.
- Add DiD to the cell suspension at a final concentration of 5 µM and vortex the suspension immediately to ensure the dye doesn't precipitate out of solution and fail to label the cells.
- Incubate for 10 min at 37 °C then wash by spinning at 500 x g for 5 min, decanting the liquid and resuspending the pellet in an appropriate volume for IV injection (~100 – 150 µl)
- Resuspend the cells in 200 µl of PBS and collect into an insulin syringe. NOTE: an insulin syringe is recommended over a conventional syringe because it has no needle dead space and therefore allows administering the entire cell suspension to the mouse.
- Inject cells into a lethally irradiated mouse via tail vein injection. NOTE: Irradiation has known, considerable impact on the bone marrow microenvironment (both hematopoietic and stromal components), which develops over time. It is therefore very important to maintain consistent timing for irradiation, cell injection (4 to 24 hr from irradiation for best engraftment) and imaging. Here irradiation is performed 6 hr before injection and imaging is performed 20 hr after injection.
- Place the mouse in a warm chamber (37 °C) until the tail vein appears vasodilated.
- Move the mouse into the restrainer and carefully slide the needle into the tail vein.
- Inject the cells into the vein – no resistance to injection should be encountered.
- Leave the mouse O/N in order to allow the cells to migrate to the bone marrow spaces.
2. Preparing the Mouse for Imaging
- Autoclave one pair of fine forceps and one pair of fine scissors as well as a headpiece prior to use and store in a sterile environment.
- Make up anesthetic as fresh as possible, (0.38 ml Ketamine + 0.25 ml Medetomidine + 4.47 ml Water). NOTE: other approved anesthetics, including isoflurane and other injectable, will be effective too. The cocktail described is to be administered intraperitoneally at 0.1 ml per 10 g of body weight, with top-ups of 30 – 50 µl administered every 45 min to 1 hr.
- Switch on the two-photon laser if required then start the microscope and software, calibrating the stage when prompted. Switch on the lasers and allow them to warm up and stabilize while preparing the mouse for imaging.
- Anesthetize the mouse using the prepared anesthetic mixture (step 2.2) for the chosen anesthetic via intraperitoneal injection. Monitor the onset of deep anesthesia via pedal reflex now and at regular intervals throughout the protocol. NOTE: the mouse must be monitored carefully throughout the procedure and if anesthesia appears to be lightening (typically after 45 – 60 min) then administer a top-up dose (as detailed in 2.2). Expect some variation between mice of different ages and sex. It is important to discuss the anesthesia protocol with your local veterinary officer to ensure adequate precautions are taken to ensure the mouse's welfare.
- Swab the top of the scalp with 70% ethanol on a tissue, ensuring not to get any ethanol in the eyes of the mouse.
- Using sterile forceps and scissors, carefully remove the central portion of the scalp to expose the calvarium area to be imaged: make a small incision at the back of the head between the ears by lifting the skin up with the forceps. While holding the skin up, slide the scissors under the skin and gently cut along the outside of the desired imaging area.
- Wipe the exposed bone with sterile cotton bud moistened with sterile PBS to keep it moist.
- Attach the metal head piece to the skull of the mouse ready for imaging.
- Mix an adequate amount of dental cement in a weigh boat until it becomes a paste and quickly apply to the bottom surface of the headpiece that will attach to the skull.
- Before the cement sets, place the headpiece onto the skull of the mouse, making sure not to get any dental cement on the imaging area, then wait for it to set.
- Apply a small amount of Intrasite Hydrogel which keeps the skull moist.
- Attach the headpiece to the holder and secure in place using the screw, ensuring that the grooves fit within the holder notches.
- Remove the hydrogel from the skull with sterile cotton buds and clean with sterile PBS before transferring to the microscope.
- Insert the holder into the microscope stage, position the heating mat under the mouse, insert the rectal thermometer probe and secure everything in place on the stage with adhesive tape.
- Place a small drop of ophthalmic ointment on the eyes of the mouse to ensure they do not dry out while under anesthetic. NOTE: the mouse must not be left unattended at any time during the imaging process while under anesthesia.
3. In Vivo Imaging: High-resolution Stacks and Time-lapse Acquisition
- Focus on the skull via the eye pieces.
- Fill the imaging window with purified, sterile water and using a water dipping lens, lower it so that it touches the water droplet.
- Focus on the top of the skull using the microscope eyepieces, using an external lamp as the light-source.
- Position the imaging area on the central suture and move towards the rear of the head to find the coronal suture as a starting position.
- Set the microscope up to allow effective excitation and detection of the relevant fluorophores specified in the excitation and emission table. NOTE: while a Leica SP5 upright confocal with a conventional galvanometer scan head running the LAS-AF software platform is used here, the procedure can be easily replicated in other microscope/software platforms. The lens used here is the Leica HCX IRAPO L 25x W/0.95 N.A. but equivalent lenses of other manufacturers are available with suitable magnification and high N.A.
- Place the software into a mode to capture both XY images as well as a 3D Z-stack and set imaging speed to 400 Hz and resolution to 512 x 512 pixels. NOTE: the setup and hardware here result in a field of view of 620 µm and this may differ on other platforms.
- Set the software so that multiple channels/tracks are able to be captured, as well as ensuring that the collection method is set to change settings between stacks if possible or between frames at the very least. Set up 3 independent capture settings, switching off the laser illumination at the end of the acquisition of each stack. NOTE: three channels will be required for this procedure to reduce crosstalk between channels.
- Setup the settings for the first sequential scan for the two photon SHG bone signal (840 nm excitation; 400 – 440 nm emission); open the shutter for the MP laser and ensure MP gain and offset are correctly setup, laser power is 12.5 – 25% and the laser is switched on. Select an appropriate PMT as the only detector and change the color to white. Note: NDD detectors with appropriate filters should be used for the bone detection when available to achieve a brighter, deeper signal.
- Repeat channel setup procedure used for the GFP channel for a combined autofluorescence (543 nm excitation; 560 – 600nm emission) and DiD (633 nm excitation; 650 – 720 nm emission) under the second settings scan, utilizing two appropriate PMTs. Change the channel color to green for the autofluorescence and red for the DiD. Note: using green as the auto-fluorescence channel allows easier detection of DiD positive cells when the two channels are overlayed – autofluorescent cells appear as yellow/orange due to being in both channels while DiD cells appear only as red since they only appear in the DiD channel. The auto-fluorescence channel is only used for this comparison and is not used in any final analysis, however if the user wishes to, this can be changed to a different color for display purposes to avoid confusion with the GFP channel.
- Finally, setup the third and final scan for GFP (488 nm excitation; 500 – 530 nm emission); make sure the shutter is open if required and then set the laser power of the 488 nm laser line to around 15% or an appropriate level for the laser being used, select an appropriate PMT as the only active detector and change its pseudocolor to green.
- Activate a 'Live' imaging mode to begin a preview scan of the selected channel and adjust detector Gain and Offset for optimal exposure. Repeat this for each scan in the sequential window.
- Save the settings of these multi-channel scans for easy reuse. NOTE: this allows the user to reload the settings in subsequent sessions.
- Activate multi-position capture, referred to as 'Mark and Find' in the demonstrated software, and reset the coordinate points.
- Scan the bone imaging area, starting from the intersection between the central and coronal suture, scan the depth of bone marrow area using both autofluorescence (pseudo colored green) and DiD (pseudo-colored red) with a composite view. NOTE: autofluorescent cells will be present in both channels and appear orange/yellow in the composite image whereas DiD labeled cells will appear as red-only cells in the composite image.
- When a cell is detected, mark this as a new coordinate position (x, y and z) in the 'Mark and Find' tool by clicking on the “Add New Position icon on the left hand side of the “Mark and Find” window.
- When the current field of view has been examined, move the stage the distance of one field of view either left/right/down/up. Repeat this process for each field of view, gradually working around the whole imaging window area to the left of the central suture, moving towards the nose of the mouse. Once the central suture bifurcation is in sight, move to the right side of the suture and repeat the procedure scanning the left side in the reverse direction (i.e., towards the coronal suture). Mark the coordinates of any new cells of interest using the 'Mark and Find' tool.
- Once scanning of the imaging window is complete, use the 'Mark and Find' tool to review the points (select the desired point and review each point individually). Set the top and bottom of a Z-stack around each cell for each position, (many software platforms will allow you to perform independent z-stacks for each position, which reduces capturing empty space). For each point, focus up and down and set top and bottom Z positions for 3D z-stack capture and update the individual point in the 'Mark and Find'. Set Z-Stack interval to 5µm and averaging for each sequential scan channel to an appropriate quality: Line or Frame average. NOTE: Increasing the number of scans to average increases the length of acquisition time.
- Acquire a high-quality reference stack for each point of interest by starting the entire scan procedure (often referred to as 'Start' rather than “Capture”). NOTE: this scan can act as a reference for future high-speed imaging.
- Once finished with the high-quality scan, activate a time-lapse setting for the capture.
- In the time-lapse settings, set the time interval to 5 min and overall run time to the desired length (e.g., 5 hr). 'Apply' these settings to the overall scan. NOTE: in order to reduce this scan time to allow a 5 min time lapse interval, one may need to reduce the number of scans used for averaging, limit the resolution to 512 x 512 pixels, convert the scanning to bidirectional (with required phase correction to align both scan directions), and/or increase the scan speed to 600 Hz or more (more than 600 Hz will cause zooming of the imaging area for the demonstrated software platform). It is also important to note that while a sequential imaging mode was used to acquire a high quality image stack initially, simultaneous acquisition is used to increase speed during the time-lapse imaging – this may result in some slight cross talk between channels which places extra importance on the initial capture of the accurately separated reference stacks.
- Commence imaging (as before by clicking the 'Start' button in the demonstrated software). NOTE: be sure to maintain the appropriate level of anesthesia by administering further, smaller doses when needed, being careful not to overdose the mouse, regularly top up the water to ensure the objective does not dry out and monitor vital signs and heating-pad temperature throughout.
- When finished, raise the lens from the holder and then remove the holder from the microscope stage, ensuring the wires from the rectal probe and heating mat are not damaged.
- Unscrew the headpiece from the stage holder and carefully remove the mouse. Cull the mouse using cervical dislocation and remove the metal head-piece by snapping it off the skull. Save data to disk and transfer to server for later analysis. NOTE: The head piece is ideally cleaned by dipping in acetone for a few hours and finally rubbing the residual cement off.
Representative Results
The custom made, high-precision mouse holder including a calvarium imaging window allows prolonged imaging of FACS purified, labeled HSPCs injected into lethally irradiated recipient mice (Figure 1A). Typically, after injection of between 15 – 20 thousand labeled cells, 8 to 15 cells are usually recognizable within the bone marrow imaging area of the skull the following day. Here it is shown how to acquire single cell resolution 3D stacks of HSPC-containing bone marrow areas of approximately 90 – 120 µm thickness , (Figure 1B), followed by 4D time-lapse movies of the HSPCs identified (Figure 1C and movie).
Suboptimal results are obtained if the dental cement is not optimally prepared, for example the water could leak from non sealed areas, and even though it can be topped up from the top of the holder, any period of time in which the lens is not dipped into water leads to black frames. Some drift in the images is inevitable due to thermal expansion of the materials of the mouse holder, however it can be accentuated by suboptimal cement adhesion, leading to progressive detachment of the mouse, and by perturbations of the imaging set up, leading to sudden jumps of the focus during time-lapse imaging. Drift during multi-point time-lapse imaging can also be caused by inaccuracies in the stage movements when revisiting previously imaged positions. Drift and stutter artifacts in time-lapse movies can be corrected for by using image registration algorithms.
Table 1. Excitation and emission settings.
| Colour Channel | Excitation | Emission |
| GFP | 488 nm | 500 – 530 nm |
| Autofluorescence | 543 nm | 560 – 610 nm |
| DiD | 633 nm | 640 – 700 nm |
| SHG | 840 nm (MP) | 400 – 450 nm |
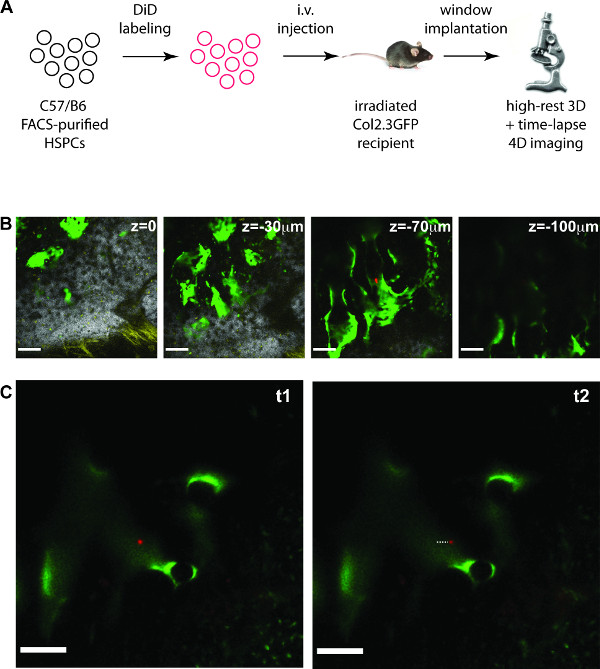
Figure 1. In vivo 4D imaging of HSPCs in mouse calvarium. (A) Outline of the experiment. (B – C) examples of results obtained. (B) 2D slices from a 3D stack (total depth is 114 μm), containing signal from collagen bone (white), DiD labeled cells (red), autofluorescent cells (yellow) and osteoblastic cells (green). Scale bars: 100 μm. (C) 2D slices from a 4D time-lapse movie showing a DiD labeled HSC (red) migrating in the proximity of osteoblastic cells (green). The dotted line highlights the displacement of the cell. Scale bars: 50 μm (B) and 100 μm (C). NOTE: The movie obtained from the time-lapse acquisition in (C) is shown during the video article. Please click here to view a larger version of this figure.
Discussion
What was accomplished?
The protocol uses time-lapse multi-dimensional imaging to monitor the migration of FACS purified, ex vivo labeled, transplanted HSPCs in mouse calvarial bone marrow. Identification of single transplanted cells has been achieved and these have been monitored over prolonged periods of time (hr) with high accuracy. Breathing induced movement artifacts have been minimized and cells have been repeatedly reimaged over a prolonged time-course.
What are future directions?
Development of the technique could progress towards recovery experiments to allow for imaging on multiple days, over the course of a week or more and use of gas anesthetics such as isoflurane to shorten and simplify the recovery process for the mouse (Note: recovery experiments require strictly sterile surgery conditions and administration of appropriate analgesics). Development of a larger color-palette of in vivo dyes as well as efficient genetic labeling of hematopoietic cells will also facilitate multi-labeling experiments, provide more insight into cell-cell interactions within the bone marrow and allow for more complex experiments in the future. Additionally, simultaneous labeling of multiple bone marrow structures and stroma components will provide a more complete picture of the events taking place in HSPC niches. Image stability of the time-lapse movies could be improved preacquisition by stabilizing the microscope further as well as minimizing temperature gradients in the sample and postacquisition by using image registration algorithms.
Limitations:
The technique described presents some limitations. Survival of mice following administration of injectable anesthetics is unpredictable and it is very easy to experience complications depending on individual response to the anesthetic. Generally, the longer an imaging session, the harder is for the mouse to recover from anesthesia and it is therefore important to carefully plan if the mouse is to be recovered, ideally limiting the duration of time lapse imaging. The use of injectable anesthetics limits the time each imaging session can last for. While it is possible to prolong the anesthesia by readministering a smaller dose of anesthetic, it is difficult perform this accurately and overdosing the mouse is one of the most frequent causes of premature termination of in vivo imaging. Gas anesthesia is less toxic and allows for a longer initial imaging session, however rapid recovery of the mouse after >2 hr long administration of isoflurane is rarely successful. Another limitation of the technique is the limited resolution of the images that can be obtained during multi-point time-lapse imaging, due to the necessity to acquire images rapidly. This, coupled with focal drift or field jumps (discussed above), can make tracking of cells difficult.
Comparison with alternative methods:
HSPCs are usually labeled with the lipophilic dye DiD, but DiR and DiI dyes are equivalent alternatives and other cell dyes can be used as long as sufficiently bright, as reviewed in[16]. Even though ex vivo labeling of cells with DiD has been shown to not affect the long-term function of HSCs, it has the limitation that DiD (or any other chemical dye) is diluted upon cell division. Since HSCs proliferate during the first days following transplantation, the signal to noise ratio for these cells rapidly deteriorates. Endogenous labeling of cells by means of genetic manipulation and expression of fluorescent proteins is a viable alternative method, as long as the fluorescent proteins are expressed at sufficiently high levels to generate signal detectable through the skull bone. There are a number of alternative techniques available to perform imaging of HSPCs within the bone marrow space, each with their own benefits and limitations. Insertion of fiber optic-based imaging devices allowed for imaging of bone marrow reconstitution in long bones[8], while confocal imaging following surgical exposure of the tibia and thinning of bone enamel with a surgical drill[12] produced detailed images of HSPCs residing in the peripheral areas of long bones. These methods however are far more invasive than the one described here and therefore do not permit to repeat imaging sessions focusing on the same bone marrow area within the same mouse. Moreover, it is possible that these techniques may affect the cells observed given the inevitable response to extensive damage in the surrounding tissue. HSPCs have been imaged for short periods of time through coverslips placed over excised, fractured femurs[11], however the impact of this harsh preparation of the tissue on HSPCs is not clear and the technique was used uniquely to obtain single time point observations. Fixed bones have been sectioned into serial sections, stained, and the obtained images reconstructed into a 3D model, providing excellent 3D resolution, especially when vascular casts are used[17], and recently Laser Scanning Cytometry (LaSC) has been used to obtain quantitative measures about HSPCs in situ, highlighted by immunostaining[18] but are unable to provide any temporal information about the cells behavior. The techniques described in this new technique provide a combination of good resolution in 3D, sufficient temporal sampling for monitoring cells in 4D and they are relatively non-invasive, therefore offering an ideal approach to achieve long-term monitoring of HSPCs interacting with the bone marrow microenvironment.
Divulgations
The authors have nothing to disclose.
Acknowledgements
Imaging was carried out on an upright Leica SP5 confocal microscope located within the FILM facility of Imperial College London, managed by Dr Martin Spitaler.
The specimen holder and headpiece was made in collaboration with the Chemical Engineering department of Imperial College London, with advice from Samuel Jones and Dr Simon Schultz.
Nicola Ruivo, Francisco Diaz and CBS staff at Imperial College were instrumental for their assistance and advice on mouse husbandry. Mark Scott is funded by HFSP and FILM, Olufolake Akinduro by CRUK and Cristina Lo Celso by CRUK, KKLF, BBSRC and HFSP.
Materials
| Name of Material/ Equipment | Company | Catalog Number | Comments/Description |
| Ketamine(Narketan 100mg/ml) | Vetoquinol | 407575 0107 C | Other anesthetics may be used in place of this. |
| Medetomidine(Domitor 1mg/ml) | Elanco | 134800-2 | Other analgesics may b used in place of this. |
| Vibrant DiD cell labeling solution | Roche Products Ltd. | V22887 | Other colors are available and may be used if required. |
| Lacrilube ophtalmic ointment | Allergan | n/a | avaliable by prescription by the local vet |
| Kemdent "Diamond Carve" glass ionomer cement | Associated Dental Products Ltd. via Kemdent | SUN527 | We use shade A3, but any shade of cement can be used. |
| PBS | multiple equivalent | n/a | |
| Sterile water | multiple equivalent | n/a | |
| Insulin syringes | Terumo Medical Corporation | SS10M3109 | 1mL, 31G |
| Mouse restrainer | Harvard Apparatus | 340031 | Cone type (small) |
| Autoclaved forceps and scissors | Agar Scientific | AGT577 AGT5034 |
Iris scissors – 90mm; Dumont HP tweezers (0.06mm tip) |
| Weigh boat and cement stirrer | VWR | 611-1996/231-0370 | 5mL weigh-boat (black)/ Wooden tongue depressor |
| Leica SP5 upright confocal microscope with motorized stage and two-photon laser | Leica Microsystems | Not available | Call for quote |
| 25x water dipping objective lens (N.A. = 0.95) | Leica Microsystems | 11506323 | HCX IRAPO L lens |
| Custom designed mouse holder | Imperial College Engineering Workshop | N/A | See Figure 1 for details |
| Heating pad with rectal thermometer probe | BASi Instrumentation | FHC-40902/ FHC-40908/ FHC-4090502 | Heat mat/ Control box/ Thermometer probe |
References
- Lo Celso, C., Scadden, D. T. The Hematopoietic Stem Cell Niche at a Glance. J Cell Sci. 124 (Pt 21), 3529-3535 (2011).
- Wang, L. D., Wagers, A. J. Dynamic Niches in the Origination and Differentiation of Hematopoietic Stem Cells. Nat Rev Mol Cell Biol. 12 (10), 643-655 (2011).
- Schofield, R. The Relationship Between the Spleen Colony-forming Cell and the Hematopoietic Stem Cell. Blood Cells. (1-2), 7-25 (1978).
- Bischoff, R. Interaction Between Satellite Cells and Skeletal Muscle Fibers. Development. 109 (4), 943-952 (1990).
- Cotsarelis, G., Sun, T. T., Lavker, R. M. Label-retaining Cells Reside in the Bulge Area of Pilosebaceous Unit: Implications for Follicular Stem Cells, Hair Cycle, and Skin Carcinogenesis. Cell. 61 (7), 1329-1337 (1990).
- Doetsch, F., Caillé, I., Lim, D. A., Garcia-Verdugo, J. M., Alvarez-Buylla, A. Subventricular Zone Astrocytes are Neural Stem Cells in the Adult Mammalian Brain. Cell. 97 (6), 703-716 (1999).
- Palmer, A. E., Tsien, R. Y. Measuring Calcium Signalling Using Genetically Targetable Fluorescent Indicators. Nature Protocols. 1 (3), 1057-1065 (2006).
- Lewandowski, D., et al. In vivo Cellular Imaging Pinpoints the Role of Reactive Oxygen Species in the Early Steps of Adult Hematopoietic Reconstitution. Blood. 115 (3), 443-452 (2010).
- Sipkins, D. A., et al. In vivo Imaging of Specialized Bone Marrow Endothelial Microdomains for Tumour Engraftment. Nature. 435 (7044), 969-973 (2005).
- Lo Celso, C., et al. Live-animal Tracking of Individual Haematopoietic Stem/Progenitor Cells in their Niche. Nature. 457 (7225), 92-96 (2009).
- Xie, Y., et al. Detection of Functional Haematopoietic Stem Cell Niche Using Real-time Imaging. Nature. 457 (7225), 97-101 (2009).
- Köhler, A., et al. Altered Cellular Dynamics and Endosteal Location of Aged Early Hematopoietic Progenitor Cells Revealed by Time-lapse Intravital Imaging in Long Bones. Blood. 114 (2), 290-298 (2009).
- Schultz, S. R., Kitamura, K., Post-Uiterweer, A., Krupic, J., Häusser, M. Spatial Pattern Coding of Sensory Information by Climbing Fibre-evoked Calcium Signals in Networks of Neighboring Cerebellar Purkinje Cells. J Neuroscience. 29 (25), 8005-8015 (2009).
- Lo Celso, C., Lin, C. P., Scadden, D. T. In vivo Imaging of Transplanted Hematopoietic Stem and Progenitor Cells in Mouse Calvarium Bone Marrow. Nat Protoc. 6 (1), 1-14 (2011).
- Lo Celso, C., Scadden, D. Isolation and Transplantation of Hematopoietic Stem Cells (HSCs). Journal of Visualized Experiments. (2), e157 (2007).
- Progatzky, F., Dallman, M. J., Lo Celso, ., C, From Seeing to Believing: Labelling Strategies for in vivo Cell-tracking Experiments. Interface Focus. 3 (3), 20130001 (2013).
- Ellis, S. L., et al. The Relationship Between Bone, Hemopoietic Stem Cells and Vasculature. Blood. 118 (6), 1516-1524 (2011).
- Nombela-Arrieta, C., et al. Quantitative Imaging of Haematopoietic Stem and Progenitor Cell Localization and Hypoxic Status in the Bone Marrow Microenvironment. Nature Cell Biology. 15, 533-543 (2013).
