To demonstrate the necessary steps required for setting up a large-scale screen, Figures 2-5 are representative control experiments related to the quality of the assay. These are essential control experiments for a consistent assay over several days of screening or for comparison of different screens.
Assessing the Fluorescence Signal-to-noise Ratio
To ensure the stability and quality of the fluorescein-labeled oligoribonucleotide substrate in a large-scale setup, Z’-factor, defined as the difference between the assay background and the maximum signal was calculated using the active ribozyme molecule (A6Rbz). Assays with Z’-factor >0.5 are considered acceptable for high-throughput screen. Figure 2 shows representative data using the FRET substrate labeled with 5’ fluorescein (FAM; emitter) and 3’ N,N’-tetramethylrhodamine (TAMRA; quencher) (A6Rbz_F/T). This substrate produced a Z’-factor of 0.64 when calculated using 72 replicates in the presence and absence of A6Rbz.
In this assay an alternative substrate using Iowa Black dark quencher (A6Rbz_F/Ib) can yield a better signal-to-noise ratio compared to TAMRA. This is because Iowa Black, unlike TAMRA, emits absorbed energy as heat and not light. The Z’-factor obtained using the FAM/Iowa Black (F/Ib)-labeled substrate was 0.68. The improvement in the Z’-factor for the F/Ib-labeled substrate over TAMRA-labeled substrate is because of its relatively lower background. These representative results show that both substrates are viable options for use in a high-throughput screen.
Determining the Editing Activities of Glycerol Gradient Fractions
To determine and select the most active editosome fractions for experiments, the glycerol gradient fractions were tested for in vitro editing using the fluorescence-based assay (Step V). These data show (Figure 3) fractions 7-12 as the most active fractions (≥50% editing activity; with the most active fraction as 100%). These fractions can be combined when more editosome is required.
Calculating the Z’-factor when the Editosome Fractions were Combined
To determine the effect of combinig the editosome fraction, the Z’-factor value of 0.6 was calculated when the most active fractions (F8+F9+F10) were used as the source of editosome. To calculate the Z’-factor 72 replicates were tested in the presence and absence of the editosome (Figure 4). The F/Ib substrate was used in this experiment. These data show that based on the Z’-factor, combining the glycerol gradient fractions have minimal effect on the quality of the assay.
Representative Control Experiment for the Assay
Representative data comparing different control samples were used to analyze the variables in the assay. As shown in Figure 5, reactions missing any RNA editing components (reactions 1-4) do not cleave the substrate. Cleavage of the substrate as measured by the fluorescence and relative editing activity is only observed in the presence of all components of the editing reactions in the absence of an inhibitor (reaction 5) or in the presence of all editing components and an inactive compound (reaction 6). In contrast, an inhibitory compound can block RNA editing and is used as a positive control (reaction 7). Here, Mordant black (MrB) and C53 compounds were used as the positive and negative controls, respectively.

Figure 1. Hammerhead ribozyme-based in vitro RNA editing assay. A) Step-by-step schematic representation of the fluorescence-based in vitro editing assay: (a) Hybridization of the pre-edited hammerhead ribozyme (pre-edited A6Rbz) with its guide RNA (gA6Rbz). (b) Recognition and interaction of the RNA duplex by the purified editosome complex. (c) Deletion RNA editing catalyzed by the editosome. (d) Dissociation of the edited A6Rbz from the editosome and gA6Rbz by heating at 85 °C and addition of guide RNA competitor (gA6Rbz_comp). (e) Hybridization of the FRET substrate with the active A6 hammerhead ribozyme (active A6Rbz). (f) Detection of FAM signals following cleavage of the FRET substrate by the active ribozyme. B) The pre-edited hammerhead ribozyme (pre-edited A6Rbz) is shown in association with gA6Rbz that specifies the deletion of three Us (double-headed arrow) from the editing site (ES). As a result of RNA editing in the presence of functional editosome the inactive ribozyme is edited into its active form (edited A6Rbz) that can now cleave the FRET substrate (cleavage site indicated by an arrow). The conserved 5′-CUGA-3′ of the edited A6Rbz in the catalytic core essential for ribozyme activity (edited site) is highlighted (This figure has been modified from Moshiri19). Please click here to view a larger version of this figure.
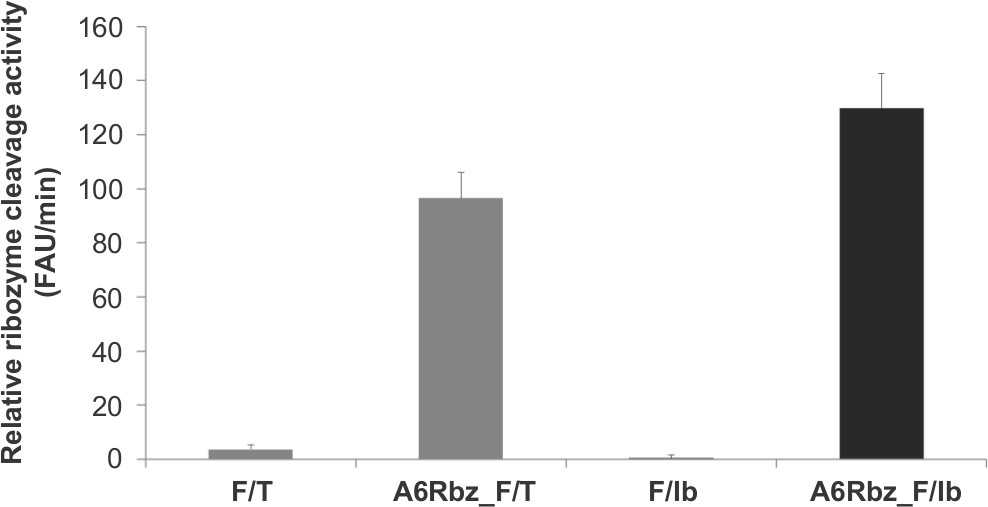
Figure 2. Signal-to-noise ratio comparison between FRET substrates harboring different quenchers. Ribozyme (A6Rbz) activity using FAM/TAMRA (F/T) and FAM/Iowa Black FQ (F/Ib) substrates. The y-axis represents fluorescence arbitrary unit (FAU) per minute.
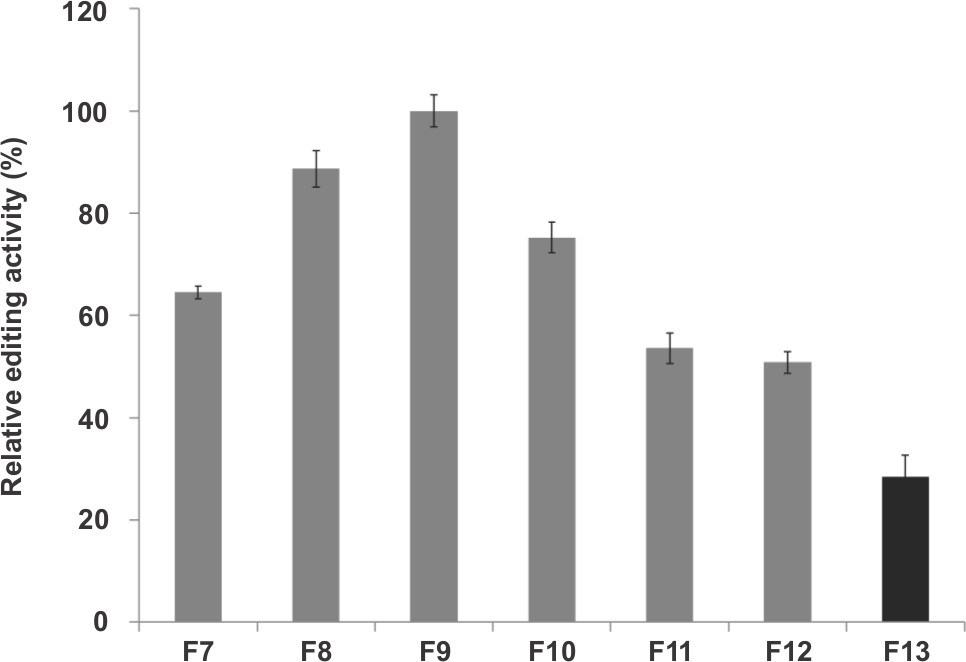
Figure 3. RNA editing activity of select fractions obtained from glycerol gradient centrifugation of the mitochondrial lysate. Fraction #9 (F9) has the highest activity. The y-axis represents relative editing activity in percentage, considering F9 as 100%.
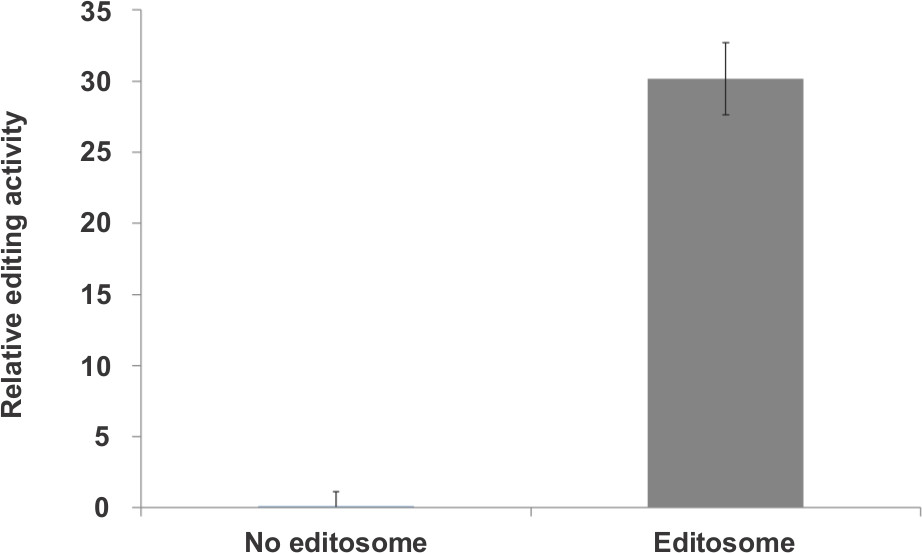
Figure 4. Z’-factor calculation using the most active fractions (F8+F9+F10) as the editosome source. Experimental variation was obtained from 72 replicates of each type of reaction. Z’-factor was calculated as 0.6. The y-axis represents relative editing activity.
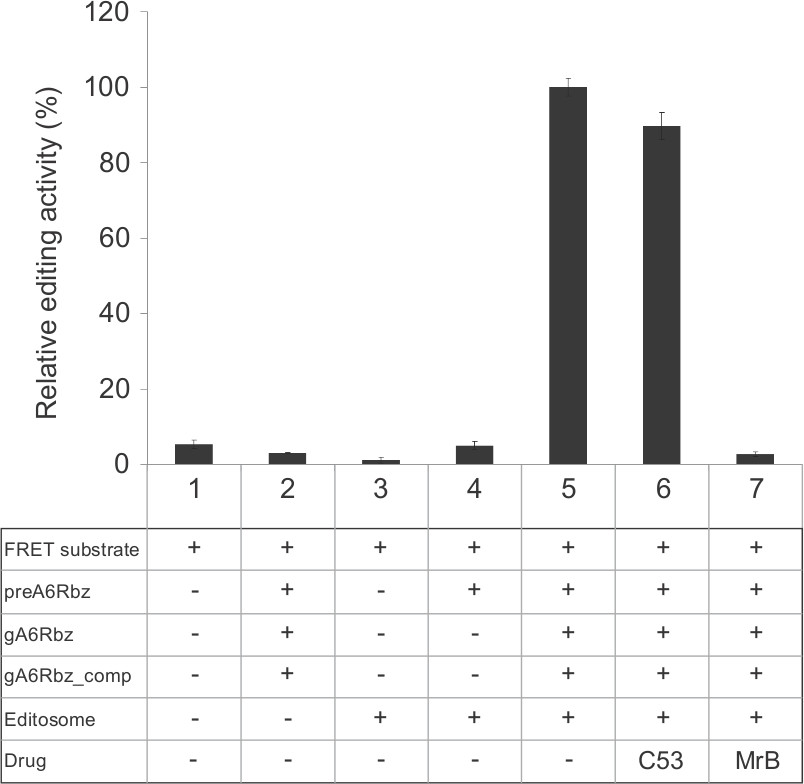
Figure 5. Representative experiment for the fluorescence-based RNA editing assay. Reactions were performed in a final volume of 20 µl per well and CFX384 TouchTM real-time PCR detection system was used for measuring the fluorescence signal. The graph presents various controls in addition to a complete editing reaction that contains all components for the assay (#5). The fluorescence was measured in the presence of FRET substrate alone (#1) to assess the integrity of the substrate. Sample #2 was performed in the absence of the editosome to monitor any ribozyme activity prior to editing. To assess the effect of denatured editosome on the substrate, the fluorescence was measured in the presence of the editosome and FRET substrate only (#3). To test the guide-directed editing specificity, a sample in the absence of gA6Rbz was used (#4). A non-inhibitory (C53, #6) and an inhibitory compound (MrB, #7) were used to test the effect on the editing assay. While RNA editing is inhibited significantly by MrB, C53 has negligible effect. The error bars correspond to an experimental variation (standard deviation) between 10 replicates. The y-axis represents relative editing activity in percentage, considering the complete reaction (#5) as 100%.
| 10% | 30% | |
| 2x HHE gradient buffer | 5 ml | 5 ml |
| Glycerol | 1 ml | 3 ml |
| DEPC H2O | 4 ml | 2 ml |
| 1 M DTT | 10 µl | 10 µl |
Table 1. 10% & 30% Glycerol Solution (10ml each).
| Pre-edited A6 Ribozyme (preA6Rbz) | |
| preA6Rbz DNA template | 5’-ACATTTGATCTATTGTTTCGTCCTCACGGACTCA TCAAAAGTCACAACTTTCCCTTTCTCTCCTCCCCCTAACCTTT CCCCCTATAGTGAGTCGTATTA-3’ |
| preA6Rbz RNA | 5’-GGGAAAGGUUAGGGGGAGGAGAGAAAGGGAAA GUUGUGACUUUUGAUGAGUCCGUGAGGACGAAACAAUAGA UCAAAUGU-3’ |
| RNA stock conc. | 1 μM |
| Guide A6 Ribozyme (gA6Rbz) | |
| gA6RBz DNA template | 5′-AAAAAAAAAAAAAAAAATAATTATCATATCACTGT CAAGGGAAAGTTGTGAGGGTGATGAGTCCGTGTATATCCCC CTATAGTGAGTCGTATTA-3′ |
| gA6Rbz RNA | 5′-GGGAUAUACACGGACUCAUCACCCUCACAACUU UCCCUUGACAGUGAUAUGAUAAUUAUUUUUUUUUUUUUUUUU-3’ |
| RNA stock conc. | 2.5 μM |
| Guide A6 Ribozyme Competitor (gA6RBz_comp) | |
| gA6Rbz_comp DNA template | 5′-GGATATACACGGACTCATCACCCTCACAACTTTC CCTTGACAGTGATATGATAATTATTTTTTTTTTTTTTTTTCCCTAT AGTGAGTCGTATTA-3′ |
| gA6Rbz_comp RNA | 5’-AAAAAAAAAAAAAAAAAUAAUUAUCAUAUCACUG UCAAGGGAAGUUGUGAGGGUGAUGAGUCCGUGUAUAUCC-3’ |
| RNA stock conc. | 12.5 μM |
| Active A6 Ribozyme (A6RBz) | |
| A6Rbz DNA template | 5′-ACATTTGATCTATTGTTTCGTCCTCACGGACTCAT CAGTCACAACTTTCCCTTTCTCTCCTCCCCCTAACCTTTCCCC CTATAGTGAGTCGTATTA-3′ |
| A6Rbz RNA | 5’-GGGAAAGGUUAGGGGGAGGAGAGAAAGGGAAA GUUGUGACUGAUGAGUCCGUGAGGACGAAACAAUAGAUCA AAUGU-3’ |
| RNA stock conc. | 1 μM |
| FRET substrate | |
| TAMRA quencher | 5’-FAM-GAUCUAUUGUCUCACA-TAMRA-3' (Eurogentec) |
| Iowa Black quencher | 5’-FAM- GAUCUAUUGUCUCACA-Iowa Black-3' (IDT) |
| RNA stock conc. | 15 μM (for both) |
Table 2. DNA Templates and RNA Substrates.
| Composition | Editing reaction (μl) |
| 10x HHE | 1.5 |
| 0.1 M CaCl2 | 1 |
| 100 mM ATP | 0.2 |
| 10% Tritonx-100 | 0.2 |
| 500 ng/μl Torula Yeast RNA | 1 |
| Editosome | 5 |
| RNase free H2O | 7.1 |
| 1 μM preA6Rbz | 1 |
| 2.5 μM gA6Rbz | 1 |
| Total | 18 |
Table 3. Reaction Composition and Master Mix.
