For all positive-strand RNA viruses, the reliability and efficiency of a reverse genetics system depend on the genetic stability of a cloned full-length cDNA, whose sequence is equivalent to the consensus sequence of viral genomic RNA.27 Figure 1 shows a five-step strategy for the construction of a full-length infectious cDNA as a BAC for JEV SA14-14-228: Step 1, purification of viral RNA from the cell culture supernatant of JEV-infected BHK-21 cells (Figure 1A); Step 2, synthesis of four overlapping cDNA amplicons (F1 to F4) spanning the whole viral genome (Figure 1B); Step 3, subcloning of each of the four contiguous cDNA fragments into a BAC vector, creating pBAC/F1 to pBAC/F4 (Figure 1C); Step 4, modification of the cloned cDNAs for in vitro run-off transcription with SP6 RNA polymerase, i.e., placing an SP6 promoter sequence immediately upstream of the viral 5'-end (pBAC/F1SP6), eliminating a pre-existing internal Xba I site at nucleotide 9131 by introducing a silent point mutation, A9134→T (pBAC/F3KO), and inserting a new artificial Xba I run-off site immediately downstream of the viral 3'-end (pBAC/F4RO) (Figure 1D); and Step 5, assembly of a full-length SA14-14-2 cDNA BAC, pBAC/SA14-14-2 (Figure 1E). Table 1 lists the oligonucleotides used in this cloning procedure.28
For the construction of a functional JEV cDNA, the first important step is the synthesis of the four overlapping cDNA fragments using the purified viral RNA as a template for RT-PCR. Figure 2 provides a representative result for the four RT-PCR products that were electrophoresed on a 0.8% agarose gel. This gel demonstrates clearly that a full-length JEV cDNA is amplified into four overlapping cDNA fragments. Occasionally, RT-PCR reactions might yield one or more additional virus-specific or nonspecific products that are mostly smaller than the expected product, because of the nonspecific annealing of primers during cDNA synthesis/amplification. On the other hand, little or no expected RT-PCR product would be amplified because of accidental RNase contamination during the viral RNA isolation or improper RT-PCR performance.
The next key step is the cloning and modification of a partial- or full-length JEV cDNA in BAC, which is a relatively straightforward procedure that uses standard recombinant DNA techniques.69 Figure 3 presents a representative outcome for the purification of the BAC clone containing a full-length cDNA of JEV SA14-14-2 by banding in a CsCl-EtBr gradient. In this experiment, after centrifugation for 16 hr at 401,700 × g, two distinct bands, i.e., the E. coli chromosomal DNA above and the supercoiled BAC plasmid DNA below, are visible in the middle of the tube under long-wave ultraviolet light. A minimal volume (~400 µl) of the lower BAC DNA band was carefully collected by poking a hole with a syringe on the side of the tube. Subsequently, the EtBr was extracted from the BAC DNA by butanol extraction, and the EtBr-free BAC DNA was concentrated by ethanol precipitation.
The final step is the determination of the specific infectivity of the synthetic RNAs transcribed in vitro from the full-length SA14-14-2 BAC (pBAC/SA14-14-2) after RNA transfection into permissive cells (Figure 4). This step involves three sequential steps: Step 1, linearization of the full-length SA14-14-2 cDNA at the 3'-end of the viral genome (Figure 4A); Step 2, production of synthetic RNAs from the linearized cDNA by run-off transcription (Figure 4B); and Step 3, rescue of the recombinant viruses in BHK-21 cells transfected with the synthetic RNAs (Figure 4C). Experimentally, two independent clones of pBAC/SA14-14-2 were linearized with Xba I digestion and treated with MBN to remove the four-base 5' overhang generated by the Xba I digestion. The linearized BACs were cleaned up by phenol-chloroform extraction, followed by ethanol precipitation. The linearization of the two purified BACs was demonstrated on a 0.8% agarose gel (Figure 5A). The phenol-chloroform extraction must be done carefully to ensure that the linearized BACs are RNase-free. Each of the two linearized BACs served as a cDNA template for run-off transcription using SP6 RNA polymerase in the presence of the m7G(5')ppp(5')A cap analog. The integrity of the synthetic RNAs was shown by running aliquots of the two transcription reaction mixtures on a 0.6% agarose gel, along with a reference 1 kb DNA ladder (Figure 5B). In this simple assay, the major prominent RNA band always migrated just below the 3 kb reference DNA band and appeared to be sharp. However, degraded RNA would have a smeared appearance on the same gel.
An infectious center assay is the gold standard for determining the specific infectivity of the synthetic RNAs. This assay was done by electroporating BHK-21 cells with RNA samples, seeding equal aliquots of the 10-fold serially diluted electroporated cells in 6-well plates containing naïve BHK-21 cells (3 × 105 cells/well), and overlaying agarose onto the cell monolayers. After incubation for 4 days, surviving cells were fixed with formaldehyde and stained with a crystal violet solution to quantify the number of infectious centers (plaques), which corresponds to the number of infectious RNA molecules delivered into the cells (Figure 6A). Since the cDNA template used for in vitro transcription has been proven to be non-infectious,27 an aliquot of the transcription reaction mixture was directly used for electroporation. Electroporation is the preferred method for RNA transfection; alternatively, RNAs can be transfected by other methods using DEAE-dextran and cationic liposomes. RNA electroporation is very effective, but "arcing" of the electric pulse occurs rarely if salts are present in the electroporation reaction or if the electroporation cuvette is reused. The expression of viral proteins in RNA-transfected cells was examined by immunofluorescence assays using an anti-NS1 rabbit antiserum (Figure 6B). The production of viral particles accumulated in the supernatants of RNA-transfected cells was analyzed by plaque assays (Figure 6C). The results of these experiments show clearly that the cDNA-derived synthetic RNAs are infectious in permissive BHK-21 cells, generating a high titer of recombinant viruses.
| Oligonucleotide | Sequencea (5' to 3') | Posteb | Polarity |
| 1RT | TAGGGATCTGGGCGTTTCTG GCAAAT |
2578–2603 | Antisense |
| 1F | aatcccgggAGAAGTTTATC TGTGTGAACTT |
1–22 | Sense |
| 1R | attgcggccgcCCACGTCGT TGTGCACGAAGAT |
2532–2553 | Antisense |
| 2RT | TTCTGCCTACTCTGCCCCTC CGTTGA |
5975–6000 | Antisense |
| 2F | aatcccgggTCAAGCTCAGT GATGTTAACAT |
1800–1821 | Sense |
| 2R | attgcggccgcGATGGGTTT CCGAGGATGACTC |
5929–5950 | Antisense |
| 3RT | ACGGTCTTTCCTTCTGCTGC AGGTCT |
9426–9451 | Antisense |
| 3F | aatcccgggGAGGATACATT GCTACCAAGGT |
5500–5521 | Sense |
| 3R | attgcggccgcGTAAGTCAG TTCAATTATGGCT |
9380–9401 | Antisense |
| 4RT | AGATCCTGTGTTCTTCCTCA CCACCA |
10952–10977 | Antisense |
| 4F | aatcccgggAGTGGAAGGCT CAGGCGTCCAA |
9200–9221 | Sense |
| 4R | attgcggccgcAGATCCTGT GTTCTTCCTCACC |
10956–10977 | Antisense |
| SP6F | cataccccgcgtattcccac ta |
Sense | |
| SP6R | ACAGATAAACTTCTctatag tgtcccctaaa |
1–14 | Antisense |
| F1F | aggggacactatagAGAAGT TTATCTGTGTG |
1–17 | Sense |
| F1R | TGGATCATTGCCCATGGTAA GCTTA |
638–662 | Antisense |
| X1F | CGAATGGATCGCACAGTGTG GAGAG |
8403–8427 | Sense |
| X1R | AAAGCTTCAAACTCAAGATA CCGTGCTCC |
9120–9148 | Antisense |
| X2F | GGAGCACGGTATCTTGAGTT TGAAGCTTT |
9120–9148 | Sense |
| X2R | cacgtggacgagggcatgcc tgcag |
Antisense | |
| ROF | CCAGGAGGACTGGGTTACCA AAGCC |
10670–10694 | Sense |
| ROR | agggcggccgctctagAGAT CCTGTGTTCTTCCTCACCAC |
10954–10977 | Antisense |
| aJEV sequences are shown in uppercase letters, and BAC sequences are indicated in lowercase letters. bNucleotide position refers to the complete genome sequence of JEV SA14-14-2 (Genbank accession number JN604986). |
|||
Table 1: Oligonucleotides used for cDNA synthesis, PCR amplification, and BAC mutagenesis.
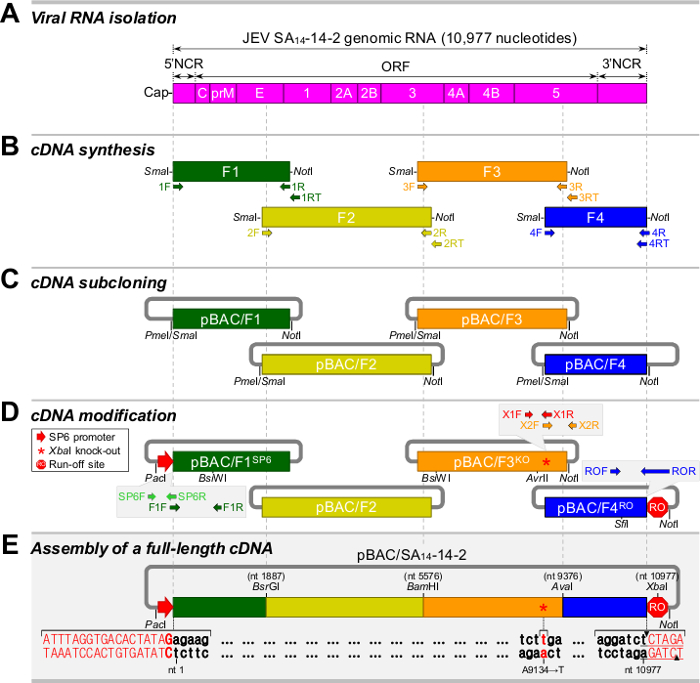
Figure 1. Strategy for the construction of a full-length cDNA of JEV SA14-14-2 as a BAC. (A) Isolation of viral RNA from JEV particles. Shown is a schematic diagram of the genomic RNA of JEV SA14-14-2. (B) Synthesis of four overlapping cDNA fragments (F1 to F4) covering the entire viral genome. (C) Subcloning of four overlapping cDNA fragments into a BAC vector, creating pBAC/F1 to pBAC/F4. (D) Modification of the cloned cDNAs for run-off transcription in vitro. pBAC/F1SP6 is a derivative of pBAC/F1 that contains the SP6 promoter sequence upstream of the viral 5'-end. pBAC/F3KO is a derivative of pBAC/F3 that contains a silent point mutation (A9134→T, asterisk). pBAC/F4RO is a derivative of pBAC/F4 that contains an artificial Xba I run-off site downstream of the viral 3'-end. (E) Assembly of a full-length SA14-14-2 BAC (pBAC/SA14-14-2). Please click here to view a larger version of this figure.
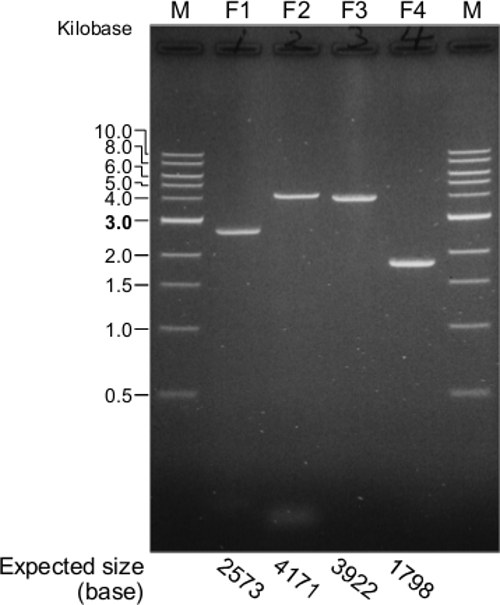
Figure 2. Synthesis of four overlapping cDNA fragments (F1 to F4) spanning the full-length genomic RNA of JEV SA14-14-2. The four RT-PCR products are evaluated by electrophoresis in a 0.8% agarose gel. M, 1 kb DNA ladder. The expected sizes of the four cDNA fragments are indicated at the bottom of the gel image. Please click here to view a larger version of this figure.
Figure 3. Purification of the BAC containing a full-length cDNA of JEV SA14-14-2. The BAC plasmid is isolated from E. coli DH10B by the SDS-alkaline lysis method and further purified by banding in a CsCl-EtBr gradient. Presented is an example of the CsCl-EtBr gradient using a 16 × 76 mm sealable polypropylene tube. Please click here to view a larger version of this figure.
Figure 4. Overview of the recovery of infectious viruses from a full-length JEV SA14-14-2 cDNA assembled in a BAC. (A) Linearization of the cDNA template. The full-length JEV BAC is cut with Xba I and treated with MBN. (B) Synthesis of the RNA transcripts. The linearized cDNA is transcribed by SP6 RNA polymerase in the presence of the m7G(5')ppp(5')A cap analog. (C) Recovery of the synthetic JEVs. The in vitro transcribed RNAs are transfected into BHK-21 cells by electroporation, which generates a high titer of synthetic virus. Please click here to view a larger version of this figure.
Figure 5. Synthesis of the RNAs by in vitro transcription using a full-length JEV BAC as a cDNA template. (A) Generation of the linearized full-length JEV BAC, pBAC/SA14-14-2. Two independent clones of pBAC/SA14-14-2 (Cl.1 and Cl.2) are linearized by digestion with Xba I and subsequent treatment with MBN. The linearized BACs are examined by electrophoresis in a 0.8% agarose gel. (B) Production of the synthetic RNAs by run-off transcription. Each of the two linearized BACs is used as a template for SP6 RNA polymerase run-off transcription. Aliquots of the two transcription reactions are run on a 0.6% agarose gel. M, 1 kb DNA ladder. Please click here to view a larger version of this figure.
Figure 6. Specific infectivity of the synthetic RNAs transcribed from a full-length JEV BAC and the recovery of synthetic virus. BHK-21 cells are mock-electroporated (Mock) or electroporated with the RNA transcripts derived from each of the two independent clones of the full-length JEV BAC (Cl.1 and Cl.2). (A) RNA infectivity. The cells are overlaid with agarose and stained with crystal violet at 4 days post-transfection. RNA infectivity is determined by infectious center assays to estimate the amount of infectious RNA electroporated into the cells (left panel). Also, representative images of infectious centers are shown (right panel). (B) Protein expression. The cells are cultured in 4-well chamber slides. Viral protein expression in RNA-electroporated cells at 20 hr post-transfection (hpt) is analyzed by immunofluorescence assays using a primary anti-NS1 rabbit antiserum and a secondary Cy3-conjugated goat anti-rabbit IgG (red). The nuclei are counterstained with 4',6-diamidino-2-phenylindole (blue). The immunofluorescence images are overlaid on their corresponding differential interference contrast images. (C) Virus yield. The cells are cultured in 150 mm culture dishes. The production of infectious virions accumulated in the culture supernatants of RNA-electroporated cells at 22 and 40 hpt is examined by plaque assays. Please click here to view a larger version of this figure.
