Herein, we show how time-lapse video microscopy is a powerful technique that can be used to follow molecular events of fluorescently-tagged proteins in a single cell. The representative results also show how this technique allows for the acquisition of high quality images. When images of the molecular process, are obtained, we have the opportunity to analyze them in different ways. Here, we analyze the interval of time between initiation of the induced mitophagy process, which is the crucial molecular process that maintain the cellular homeostasis selectively removing the damaged mitochondria.
This technique founds a broad range of applications. It can be used either for monitoring macro cellular process such as cell migration or micro molecular events through the labeling of the key molecules involved in the process that has to be studied.
Parkin-mediated Mitophagy
The selective Parkin-mediated removal of damaged and dysfunctional mitochondria process is summarized in Figure 1, where a membrane depolarization induced by the uncoupling agent FCCP triggers selective Parkin-mediated mitophagy, recruiting PINK1 to the outer mitochondrial membrane. Following, Parkin is recruited to the damaged mitochondria and interacts with PINK1. This complex Parkin-PINK1 leads to the ubiquitination of the damaged organelle and its incorporation into an autophagosome, which fuses with a lysosome. This process known as mitophagy is considered to be a crucial metabolic process in order to keep the cellular environment healthy and allow the regeneration of functional mitochondria 8,15.
Experimental Protocol
Figure 2 shows a schematic representation of the experimental protocol and the microscope set-up used for following the recruitment of EYFP-Parkin before and after FCCP-induced mitophagy process.
Parkin Recruitment to Depolarized Mitochondrial Membrane
In Figure 3A, we show Parkin distribution prior and post FCCP administration. During the basal time points (BT5, BT10 and BT15 min) EYFP-Parkin (Green) is homogeneously diffused throughout the cell and the mitochondria network (Red) appears to be well interconnected. Following FCCP administration (FT = 0 min) (Figure 3B), we observe mitochondria fractionation (FT = 5 min). However, EYFP-Parkin is still homogeneously diffused though out the cytoplasm. Mitophagy is stimulated at 55 min post FCCP administration, EYFP-Parkin is recruited at the damaged mitochondrial membranes to trigger the mitophagy process (FT = 55).
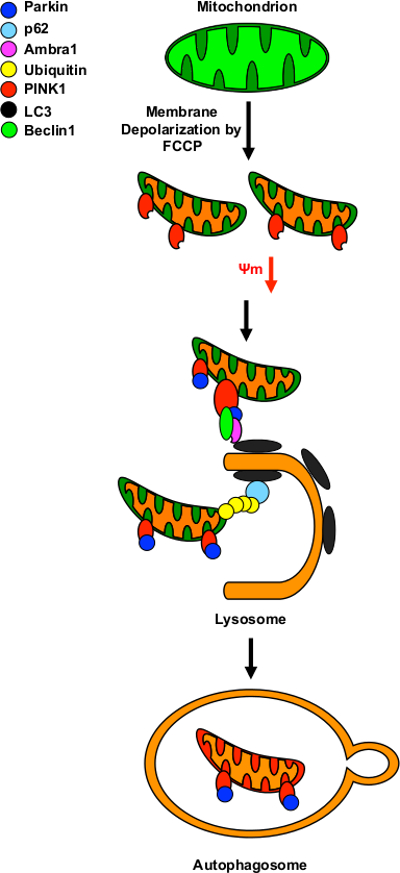
Figure 1. Schematic Representation of Parkin-mediated Mitophagy. A decreasing of membrane potential (Ψm) occurs in damaged or dysfunctional mitochondria and can be induced by the uncoupling agent FCCP. Loss of membrane potential triggers selective Parkin-mediated mitophagy, recruiting PINK1 (Red) to the outer mitochondrial membrane. Parkin (Blue) is recruited to the damaged mitochondria by directly interacting with PINK1. The presence of Parkin leads to the ubiquitination (Yellow) of the damaged organelle and its incorporation into an autophagosome, which fuses with a lysosome. Please click here to view a larger version of this figure.

Figure 2. Microscope Set-up and Representation of the Experimental Protocol. (A) Overview of the experimental events described in the protocol section. The reported experimental events allow the operator to transiently co-transfect the cells with EYFP-Parkin and pDsRed2-Mito by electroporation in order to follow the FCCP-induced mitophagy process using a (B) set-up for time-lapse video microscopy approach. BT = basal time point; FT = FCCP time point. Please click here to view a larger version of this figure.
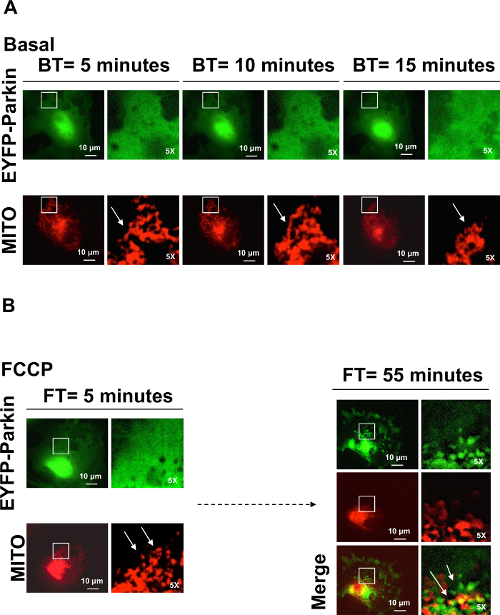
Figure 3. Fibroblast Cell Showing Parkin Recruitment after Mitochondrial Membrane Depolarization Induced by FCCP. EYFP-Parkin (green) translocation into the mitochondria (red) was monitored by time-lapse video microscopy during the basal time point (BT) and post FCCP administration (FT). From BT = 0, EYFP-Parkin and mitochondrial fluorescence were detected every 5 min. (A) EYFP-Parkin is homogenously diffused across the whole basal period and the mitochondrial network is intact (white arrows). (B) Mitochondrial fractionation due to membrane depolarization by FCCP is recorded at FT = 5 min (white arrows) and EYFP-Parkin recruitment to damaged mitochondrial membranes started at FT = 55 min (white arrows). The white boxes represent the magnified area of the panels (5X). For more details concerning the application of this experimental protocol refer to reference8. (Scale bar: 10 µm). Please click here to view a larger version of this figure.
