Cultivate Primary Nasal Epithelial Cells from Children and Reprogram into Induced Pluripotent Stem Cells
Summary
This publication demonstrates methods for successful sampling and culture of nasal epithelial mucosa from children, and reprogramming these cells to induced Pluripotent Stem Cells (iPSCs).
Abstract
Nasal epithelial cells (NECs) are the part of the airways that respond to air pollutants and are the first cells infected with respiratory viruses. They are also involved in many airway diseases through their innate immune response and interaction with immune and airway stromal cells. NECs are of particular interest for studies in children due to their accessibility during clinical visits. Human induced pluripotent stem cells (iPSCs) have been generated from multiple cell types and are a powerful tool for modeling human development and disease, as well as for their potential applications in regenerative medicine. This is the first protocol to lay out methods for successful generation of iPSCs from NECs derived from pediatric participants for research purposes. It describes how to obtain nasal epithelial cells from children, how to generate primary NEC cultures from these samples, and how to reprogram primary NECs into well-characterized iPSCs. Nasal mucosa samples are useful in epidemiological studies related to the effects of air pollution in children, and provide an important tool for studying airway disease. Primary nasal cells and iPSCs derived from them can be a tool for providing unlimited material for patient-specific research in diverse areas of airway epithelial biology, including asthma and COPD research.
Introduction
Induced pluripotent stem cells from human samples (hiPSCs) are a fast developing technology of stem cell research. They offer an alternative to embryonic stem cell (hESC) research with far fewer ethical and moral drawbacks 1,2. Although they are not epigenetically identical to hESCs 3-5, hiPSCs offer a unique way to model development and disease phenotypes, and they can be derived from tissues relevant to the disease state 5-8. New methods of generating hiPSCs are constantly being explored to identify optimal cell types to start with, as a way to prepare GMP-quality iPSCs suitable for transplantation, and also to increase the timeliness and efficiency of the reprogramming process 6,9-11.
Airway epithelial cells are critical in the development of allergic inflammation 12, and the epithelium is a major driver of allergic responses and airway remodeling through interaction with immune and stromal cells. The airway epithelium plays an essential role in the origin and persistence of lung diseases such as asthma. However, lower airway epithelial cells are difficult to obtain in a clinical setting, especially from healthy control patients and children. Data from several studies support the premise that epithelial cells from nasal mucosa are a valid and practical proxy for lower airway epithelial cells 13-20, especially when studying responses to air pollutants and allergens. The nasal mucosa consists of more than 90% ciliated airway epithelial cells and sampling these nasal epithelial cells (NECs) can be readily performed in children as young as age four or five, as it is less invasive than other cell/tissue sampling techniques and is associated with minimal risk of adverse events such as infection 20-23. It offers a rapid and simple way to sample both healthy and diseased children without long, unnecessary, and often painful bronchoscopy procedures that necessitate sedation. Previous studies have found that disease subtypes related to asthma severity can be distinguished in both the nasal mucosa as well as bronchial cell samples taken from asthmatic children, and gene expression between the two tissue types was similar in about 90% of non-ubiquitous genes 22,24. As a source for iPSCs, NECs offer advantages over other frequently utilized cell types. Fibroblasts are often used for iPSC generation, but although these cells can easily be cultured from a skin biopsy, this process typically requires local anesthesia, an incision, and sutures, and is associated with some risk of infection. Therefore, obtaining informed consent from patients for this type of biopsy can be difficult 25. One alternative to fibroblasts is peripheral blood mononuclear cells (PBMCs). However, it may be difficult to obtain sufficient blood for iPSC generation from pediatric patients. In addition, there are limitations of downstream applications for fibroblast and blood cell derived iPSCs, especially their differentiation capacity to certain cell types 5,26. Therefore, given the relative accessibility and the low risk of side effects following their collection, NECs represent an ideal cell source for iPSC generation from pediatric populations.
iPSCs have received a lot of attention recently as a platform for studying human development, generating novel disease models, and as a potential source of cells for personalized therapies. Before the full potential of this technology can be realized, the molecular underpinnings of the reprogramming process need to be elucidated, but for now this protocol and the procedures outlined within will elucidate the research studies focused on airway exposures, as well as provide a platform for studying the effects of personalized medicine involving iPSCs.
The collaborative work of several labs has led to the generation of a successful technique for not only sampling the nasal mucosa, but also culturing NECs, and reprogramming these cells to iPSCs 23. This article provides an outline of a protocol for optimal sampling, culturing, and reprogramming conditions.
Protocol
The following protocol follows the guidelines of the institutions human research ethics committee.
1. Sampling the Nasal Mucosa
NOTE: Obtain samples from subjects who are free of signs of respiratory viral infection.
- Prepare a 15 ml conical fresh before the participant visit and add 2 ml BEGM (Bronchial Epithelial Cell Growth Medium) plus 20 µl sterile Penn/Strep/Fungizone (P/S/F) (0.01%).
- Open cytology brush just before taking the sample, and be sure to keep brush sterile and do not touch it to any surfaces besides the nasal surfaces.
- Ask participant to sit still and tilt head up towards ceiling. Have smaller or more restless children sit on their hands and/or lying down to avoid swatting brush away.
- Aim brush at the back of the nose where the passage narrows (Looks like a small black hole). Slide brush down and twist wrists as the brush is removed from nostril.
- Place brush in conical and submerge in BEGM. Cut off excess brush and replace cap on tube. DO NOT VORTEX.
NOTE: If the participant is willing, obtain a second brush in the same manner but in the second nostril. Obtaining a brushing of both nostrils will result in a higher likelihood that the sampling will be successful. Sampling the same nostril may result in bleeding and will not likely improve the sample. - Keep samples as close to 37 °C as possible during transport using a beaker with warm water.
2. Cell Count and Cytospin
- Gently agitate the brush in BEGM and take 10 µl of the sample for a cell count using a hemacytometer. Add 10 µl of a live/dead stain and count only live cells under a microscope.
- Take 50 µl of the sample and dilute to 200 µl with 1x PBS. Add to cytospin funnel attached to glass slide and spin at 300 rpm for 2 min. Let slide dry O/N and stain slide following manufacturer's instructions. Allow to dry O/N, preferably in the dark.
- Stain Slide with Hema 3 Stain
- Transfer each solution (Hema fixative, light blue; Hema 3 Solution I, pink; and Hema 3 Solution II, deep blue) into a staining dish; Keep covered when not in use. Insert slides into slide staining boat
- Dip slides continuously in fixative for 30 sec, then allow excess to drain. Dip slides continuously in Solution I for 30 sec, then allow excess to drain. Dip slides continuously in Solution II for 30 sec, then allow excess to drain
- Rinse with deionized water until water runs clear. Allow to dry O/N, preferably in the dark
- Look at slide under microscope and count cells, determine percentage of epithelial cells. Should be 90% or greater if sampling is good.
3. Seeding Cells
NOTE: Carry out all cell culture procedures in a proper and certified tissue culture hood using sterile technique.
- Coat plates with Bovine Dermal Collagen (BDC), type 1. Add 0.5 ml of 3 mg/ml BDC diluted in 49.5 ml 1x PBS. Prepare less if only a few plates will be used. Coat a T25 flask with 2 ml BDC and incubate in sterile hood O/N or at 37 °C for 2 hr, if necessary.
- After incubation, aspirate any remaining liquid. Expose flasks to UV light for 30min in sterile hood. Store flasks for up to one month at 4 °C, but bring them to RT or 37 °C before seeding cells.
- Keep brush and cells in BEGM at 37 °C until ready to seed. Gently swish brush inside conical, but do not vortex.
- Plate 7 x 105 cells in a T25 flask. If there is less, use a smaller container, such as one well of a 6-well plate. This size plate requires about 3 x 105 cells.
NOTE: If there are not this many cells, it is not advisable to continue.
- Plate 7 x 105 cells in a T25 flask. If there is less, use a smaller container, such as one well of a 6-well plate. This size plate requires about 3 x 105 cells.
- Under a sterile hood, take brush out of tube and gently rotate brush on the surface of the coated flask, being careful not to scratch the coating. Discard brush in an appropriate biohazard container.
- Slowly pipette the remaining cells in BEGM out of the conical and into the T25 flask. Observe the cells under microscope. A number of floating cells will be observed and there may be some debris from the nose, which is acceptable at this stage (Figure 3A).
4. Cell Culture
- After seeding cells, let them settle for 48 hr without disruption (Figure 3). After 48 hr, slowly and gently add 2 ml warm BEGM and 20 µl sterile Penicilin/Streptomycin/Fungizone (P/S/F).
NOTE: DO NOT aspirate the original 2 ml of media. - On the 4th day after seeding, carefully aspirate all liquid, and replace with 4 ml fresh, warmed BEGM plus 1x Pen/Strep (fungizone is no longer necessary). Change media every two days and assess cells for attachment, growth, and confluence. When cells reach ~80% confluence, usually in 2-3 weeks, they are ready to be passaged.
- One T25 flask (about 2.5 x 106 cells when confluent) can be passaged into three T25 flasks or one T75 flask. After the first passage, cells should be robust and healthy, reaching confluence about every three days.
5. Passaging Cells
- Gently aspirate the media from the plate. Add 1 ml 0.05% Trypsin solution per T25 flask to the plate and place in the incubator for about 4min, or until cells detach from plate. Check the level of detachment of cells every 2 min and do not leave Trypsin on longer than 10 min.
- Add 5 ml Trypsin Neutralizing Solution (TNS), or serum-containing media to neutralize the enzyme. Remove all the liquid and cells from the flask and place in a 15 ml conical tube.
- Centrifuge at 200 x g (acceleration 0 deceleration 0) for 5 min to obtain a cell pellet. Aspirate supernatant and resuspend cells in BEGM + P/S in the volume required for the new flasks (4 ml for a T25 flask, 10 ml for a T75 flask)
- Perform cell count with a live/dead stain as described above (2.1). Add appropriate amount of BEGM and cells to the coated flasks and return to the incubator. Maintain as detailed above or cryopreserve in freezing medium (70% BEGM, 20% FBS and 10% DMSO) at a concentration of 0.5-2 x 106 cells per ml.
6. Reprogramming to iPSCs
NOTE: Before generating iPSCs, ensure adherence to all institutional regulations governing the generation and use of human iPSCs. Sterile technique is especially important for iPSC cultures, as culture medium does not routinely contain antibiotics. It is critical that NECs appear healthy and are robustly proliferative for successful reprogramming.
- Plate 5 x 105 NECs per well of a 6 well tissue culture plate. After 24 hr, add polybrene to BEGM to a final concentration of 8 µg/ml and transduce NECs by adding lentiviral particles expressing Oct4, Sox2, Klf4, c-Myc to the media. Aim to achieve a multiplicity of infection (MOI) of ~527.
- To determining the MOI, prepare serial dilutions of the concentrated-lentiviral stock and transduce HT1080 cells with these dilutions. After first unequivocally detection of dTomato expression in the HT1080 cells, calculate the number of "transducing units/ml" by scoring the number of dTomato-positive cells/small clumps of cells in each dilution.
NOTE: Typically, there will be dilutions that are too high (everything is dTomato positive) and too low (none or only a couple of positive cells). - Perform scoring from the dilutions in which discrete positive cells/small clumps of cells is identified. Determine the titer by correcting the number of transducing units/ml with the appropriate dilution factor. MOI is then the ratio of the number of transduced cells to the number of virus particles 27.
NOTE: Each dTomato positive cell/small clump is assumed to arise from a single virus particle.
- To determining the MOI, prepare serial dilutions of the concentrated-lentiviral stock and transduce HT1080 cells with these dilutions. After first unequivocally detection of dTomato expression in the HT1080 cells, calculate the number of "transducing units/ml" by scoring the number of dTomato-positive cells/small clumps of cells in each dilution.
- About 3 hr post-transduction, remove media and replace with fresh BEGM. Discard lentivirus-containing media using institutionally-approved procedures. Return transduced NECs to the incubator for 3 days.
- On day 3, replace media with fresh BEGM, and then incubate for another 3 days. Aspirate spent media, wash cells with 1x PBS and add 1 ml 0.05% Trypsin solution to the well. Passage cells with Trypsin as written above (section 5). Carefully aspirate supernatant and resuspend cells in 4 ml BEGM + P/S. Cells can be passaged to a new well of a 6 well plate coated with BDC, or added to MEF cultures, if ready (see next step).
- Prepare MEF coated plates. On day 4 add 0.1% gelatin solution (1 ml/well) to 2 wells of a 6 well (to perform +/- Thiazovivin (SPT) see step 6.6). On the next day, thaw a vial of inactivated MEFs 28.
- Place MEFs into 10 ml of MEF media (DMEM + 1x NEAA + 10% dFCS). Spin at 200 x g for 5 min to pellet. Resuspend in MEF media. Aspirate gelatin from 6 well plate and add 1.87 x 105 MEFs per well of the gelatin-coated 6 well plate. Incubate O/N at 37 °C, or for a minimum of 24 hr.
- Transfer 2 ml of BEGM containing transduced cells per well into 2 wells of a previously coated 6 well plate and incubate O/N. On the next day, replace BEGM with 2.5 ml per well of standard hESC media containing 4 ng/ml basic fibroblast growth factor. Feed cells with fresh hESC media +/- SPT daily for 10 days
NOTE: It is possible to add a cocktail of small molecules (SB431542, PD0325901, and Thiazovivin (SPT) cocktail) to enhance the efficiency and kinetics of reprogramming for NECs 23. - After ten days, continue to feed daily using hESC media without SPT. Feed cultures daily until hESC-like colonies appear (Figure 5A, P0 colony). Identify colonies harboring putative iPSCs by their high nucleus:cytoplasmic ratio and prominent nucleoli.
- Manually excise and transfer colonies with hESC-like morphology to separate wells for culture and expansion as separate lines in a feeder-free system. Excised colonies should adapt rapidly to these conditions.
- After plating excised colonies these discrete putative iPSC lines are now considered passage 1 (p1). Feed cultures daily with mTeSR1. Passage and expand iPSC lines using standard procedures. It may take several days to a week for p1 colonies to reach the size at which they should be passaged.
7. Maintaining iPSCs in Culture
- Feed and evaluate cultures daily. Manually excise areas exhibiting overt differentiation by scraping with a sterile glass pipette or micro pipette tip. Change media after daily picking.
- Passage cells after approximately every four days: Gently aspirate media, add 1 ml warm Dispase (1 mg/ml) per well of a 6-well plate (half the feeding volume), then incubate at 37 °C for about 4 min. The edges of the colonies will begin to lift, which looks like a white outline around the colony. Gently aspirate the Dispase and wash 3x with warmed DMEM/F12.
- Add 2 ml mTeSR1 and use a cell lifter to lift the colonies from the plate. Use a 5 ml serological pipette or P1000 micropipette to gently triturate the cells until the colonies are broken into smaller clusters.
- Avoid producing very small clusters or single cells, as survival and subsequent proliferation will be sub-optimal. When re-plating colonies, a 1:4 to 1:6 split will maintain optimal colony density. Gently remove colonies and media from the plate and add to new, matrigel-coated wells.
Representative Results
The initial part of the nose, called the nasal vestibule, is the area surrounded by cartilage 29. The brush needs to go smoothly past this area of the nose, beyond the nasal valve (ostium internum, or the "black hole" seen at the back of the nare), and the sample is obtained from the inferior turbinate (Figure 1). The nasal turbinates are bony structures that increase the surface area of the nose 29, making them an ideal location for sampling. The area is also lined by vascularized mucosal tissue, which closely resembles the linings of the lower airways and is also active in the immune response to environmental exposures.
Sampling the correct tissue ensures that the cells that are reprogrammed are epithelial cells. This can be evaluated by the initial cell count and cytospin, but also by observing the cells in culture. The cytospin, once spun down, dried, and stained, shows the makeup of the sample. Most samples consist of epithelial cells, which are columnar and ciliated, with one round nucleus. With the Hema 3 stain kit, they stain a light blue color with a darker purple nucleus (Figure 2), and often when taken directly from the nose they are seen clumped together. Although nasal cell samples are initially a mix of cell types as well as debris from the nose, with time in culture and with media changes, the epithelial cells become predominant (Figure 3A, B). When epithelial cells are growing to confluence, they are shaped like a freshly cracked egg, where the nucleus is large and located in the center and the cytoplasm is sort of "reaching out" and stretching as the cell prepares to divide (Figure 3B). As the cells grow together, they begin to take on more of a football shape, and fit together snugly in the dish (Figure 3C).
Once the cells are transduced, they will express the tdTomato fluorescent marker in the nucleus. Figure 4 shows the cells once they have been transduced, in bright field, fluorescence and with both images merged. Not every cell expresses the marker, and therefore not every cell was transduced, but each well should have about 90 percent transduction efficiency, and this will increase the likelihood that some cells will achieve pluripotency after being plated on MEFs.
Once co-cultured with MEFs and as cells begin to form colonies, they will gradually stop expressing tdTomato. However, this is not the best indication of colony formation. The best indication of colony formation is visual confirmation. hESC colonies, which are the "gold standard" of pluripotency, are mostly round and are composed of many small, round cells with large nuclei and a high nucleus to cytoplasm ratio. Each cell appears mostly composed of the nuclei, and other organelles are not clearly visible. Following iPSC colony harvest, and culture in standard feeder-free conditions, newly formed iPSC colonies should look much like their hESC counterparts, and the individual cells are almost identical to the hESC phenotype, as seen in Figure 5B.
Before utilizing iPSCs generated from nasal epithelial cells (NEC-iPSCs) in experiments, it is advised to assess the quality of new lines. This typically involves assessment of the karyotype, the expression of pluripotency markers, and the ability of iPSCs to differentiate into each of the 3 embryonic germ layers (endoderm, mesoderm and ectoderm). Differentiation capacity can be tested in many ways, but currently, the most stringent assay is the teratoma assay 2,23,30. More comprehensive assays such as transcriptional and epigenomic profiling are useful to determine whether any chromosomal abnormalities are introduced by reprogramming 23.
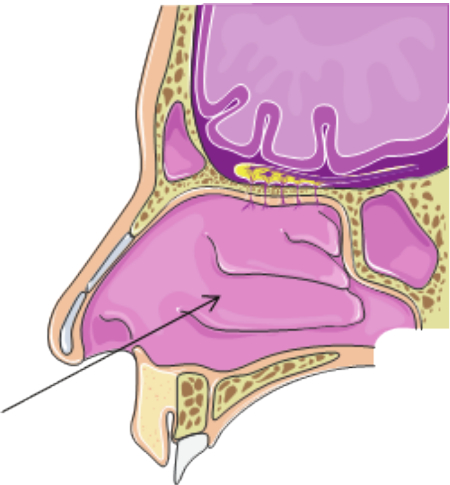
Figure 1. Inferior Turbinate. Sampling movement and the sampling target, the inferior turbinate, is indicated by black arrow. Please click here to view a larger version of this figure.
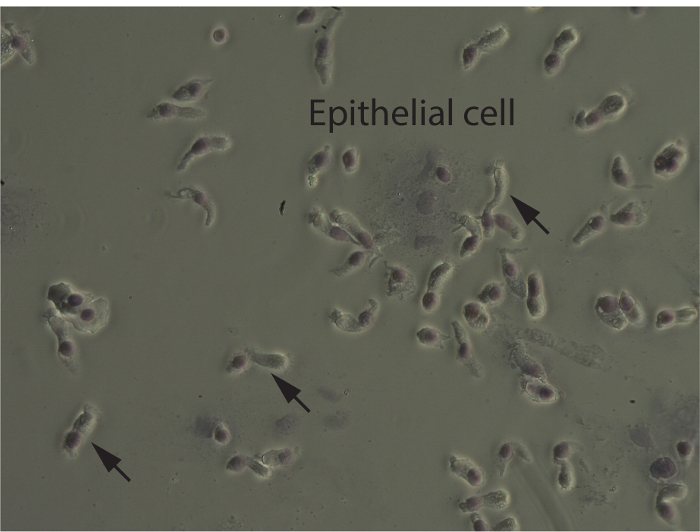
Figure 2. Cytospin. Most of the sample consists of epithelial cells, which are long and columnar, with cilia at one end and a round nucleus at the other. They are often clumped together when sampled directly from the nare. Arrows indicate individual epithelial cells. Please click here to view a larger version of this figure.
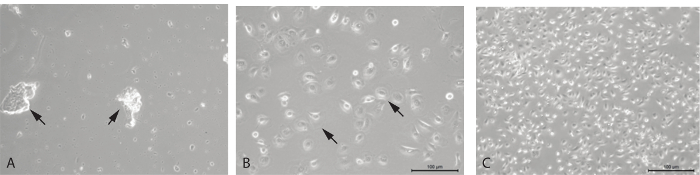
Figure 3. Primary nasal epithelial cells in culture. (A) Floating cells and debris just after sample is collected and plated. (B) Cells one day after being passaged. Cell morphology is consistent and the culture primarily consists of nasal epithelial cells, 10X magnification, and (C) 5X magnification. Please click here to view a larger version of this figure.
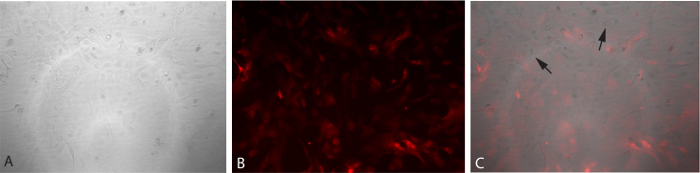
Figure 4. Transduction of NECs. Cells being transduced have taken up the vector and show the red fluorescence marker tdTomato. This marker should fade as they progress towards a pluripotent state. (A) Bright field image of transduced cells. (B) Fluorescent image of transduced cells. (C) Overlay of bright field and fluorescent images (panels A and B). Please click here to view a larger version of this figure.
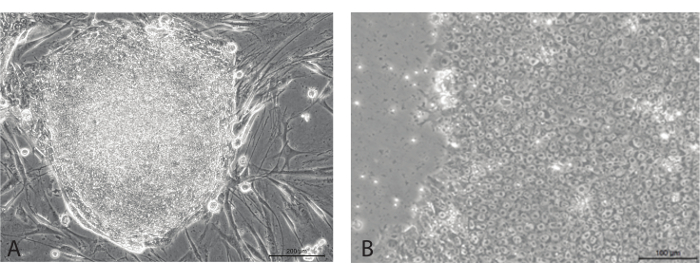
Figure 5. iPSC stem cell-like colonies. (A) Growing on MEFs, colony is mostly round on edges and shows little to no differentiation. (B) Individual cells show typical hESC-like morphology, with small round cells with large nuclei. Please click here to view a larger version of this figure.
Discussion
Nasal epithelial cells (NECs) are an accessible platform for studying airway disease, and NEC-iPSCs offer an exciting avenue to explore disease development, treatment and therapy 1,31,32. NECs can easily be obtained without stressful or potentially harmful procedures 6,23. In our experience, the sampling of the nasal mucosa as described in this protocol appears to be less stressful and better perceived than blood collection for children. Therefore, this method may be particularly useful in pediatric patient populations and relevant to studying airway diseases. The other advantage of the NECs is that once they are in culture, epithelial cells are selected for with media and culture condition and become homogenous. This is advantageous than using a complex tissue such as blood cells and makes it easier to evaluate the impact of cell origin in resulting iPSC lines using transcriptomic and epigenetic approaches.
When sampling the nasal mucosa, it is important that the correct area of the nose is sampled so that mostly epithelial cells are retrieved, as evidenced by viewing the cytospin under a microscope (Figure 2). Correct sampling will yield ≥90% epithelial cells. It is critical to obtain samples from healthy patients, and to ensure that the sample is collected appropriately in order to maximize the cell number obtained and plated. The twisting motion described in the protocol (section 1.5) ensures that the brush comes into contact with as much of the nasal surface as possible. It is a natural reflex to turn one's head when a foreign object is heading towards one's face. Therefore, in the event that a child turns his or her head or pulls away, it is essential that sampling occurs quickly and accurately. Using these techniques will increase the efficiency of every other step of the protocol, and determine the likelihood of successful reprogramming. Asking the child to lie down, and even to place their hands beneath them or have a parent or sibling hold their hands is a good way to encourage them to lie still and to help guarantee a smooth and painless sample collection.
Recently, a few other sampling techniques have been evaluated for comfort, ease, and sample outcomes, based on cell content and composition as well as RNA and DNA yields 33. The group reviewed sampling with a cytology brush versus a polyester swab, and they sampled both the inferior turbinate as well as the anterior nares by vigorously swirling the brush or swab around the nares. Sampling the inferior turbinate with a brush was found to be slightly uncomfortable, although it did yield the best RNA yields and consisted of the most ciliated epithelial cells, as compared to a polyester swab. The method proposed in this protocol is therefore verified as the most reliable method of sampling the nasal epithelia as a way to study respiratory airway epithelium.
Once the sample is obtained, it must be kept at 37 °C. One of the most effective ways is to warm a beaker of water to greater than 37 °C and nestle it into a Styrofoam container for safe transport while preparing to meet the participant. Just before the sample is collected, add cool water so that the temperature is brought to about 37 °C, and add the conical tube with the media. When the brush is added to the conical tube, return the tube immediately to the water bath and keep it there until it can be returned to the lab, at which point the tube should be put in a regulated 37 °C water bath until cells are seeded onto a culture flask. At all points, sterile technique should be used to avoid introducing foreign agents into the culture.
A considerable amount of variability amongst the samples was seen when cells were put in culture. This variability seemed to stem from both the donor source as well as the sample quality itself. One way to increase sample quality is to obtain one brush from each nostril, or two brushes per participant. After seeding the fresh sample, and as the cells are passaged, the condition of the cells should be monitored regularly to be sure they are fit to be reprogrammed. Cells that make the best candidates for are the ones that take to culture and proliferate quickly. It is not known why cells from certain donors are more robust than those from other cells, but it may be due to factors such as age, size of the nose, and health of the donor at the time of sampling. When a sample yields very few cells, or only a small number of cells attach once plated, the sample is unlikely to grow to confluence. If a struggling culture does reach confluence, the cells tend to be weak and many cells die during passaging. This type of sample is a very poor candidate for reprogramming, as it is essential that the cells be proliferating rapidly and robustly, and that they are transduced at the earliest possible passage number. It is not advisable to transfect cells that are growing slowly, whether this is due to donor variability, poor sampling, or passaging cultures too thin. If reprogramming is attempted with a poor cell culture, it is unlikely to proceed to fruition.
Preparing the cells for transduction is one part of the protocol that is most difficult to prepare for. Based on the growth of the cells, 2 x 105 to 5 x 105 cells are suggested for transduction and this may dependent on specific patient samples. Therefore it may require a few wells plated with varying cell numbers to obtain the ideal number.
This protocol outlines not only the ease of sampling, but also the relative ease and timelines of establishing primary cell cultures, and subsequent reprogramming of those primary cell cultures to iPSC lines for more sustainable and versatile research opportunities. The entire process from clinic to establishment of discrete iPSCs in culture takes about 3 months, and new methods for shortening reprogramming times are being developed 6,34,35. This field is rapidly evolving to find more efficient and complete ways to convert primary cells to iPSCs, and to subsequently differentiate iPSCs into specific cell and tissue types of interest. However, differentiation protocols for tissues important for airway disease, such as the lung epithelium, are still being worked out 36-38.
Generating iPSCs from NECs has the advantage of simple and relatively painless isolation of cells. Furthermore, it is possible that NEC-iPSCs offer advantages over iPSCs derived from other tissue types that can be exploited. The impact of iPSC epigenetics on subsequent iPSC differentiation is not completely understood, although evidence indicates that mouse and human iPSCs harbor transcriptional and epigenetic memory of their somatic cell of origin and that this selectively favors their differentiation into lineages associated with the donor cell type whilst restricting other fates 5,26. Due to the importance of the tissue of origin, and how this impacts the functional properties of iPSCs, particularly with respect to the retention of tissue-specific epigenetic marks 39-45, iPSC lines need to be more clearly studied in order to better understood the impact of the tissue of origin. It has previously been shown that NEC-derived iPSCs harbor a genomic methylation signature indicating retention of an epigenetic memory of their tissue of origin 23. Such epigenetic memory retained in NEC-iPSCs may be beneficial in differentiation of pluripotent stem cells into airway cells, as previous studies have shown that blood cell derived iPSCs exhibiting epigenetic memory of the blood cells of origin are more easily differentiated into blood cells than fibroblast or smooth muscle-derived iPSCs lacking blood cell-specific epigenetic memory 5,26. Given the enormous potential of iPSC-derived airway cells for modeling lung diseases, it will be important to study whether NECs represent an optimal cell type for differentiation of iPSCs into airway cells/tissue. In addition, this stably maintained memory may represent chromosomal locations resistant to reprogramming and candidates for disease states of donor cells. As protocols to convert iPSCs to proximal and distal lung cell types are developed 36-38, these NEC-iPSCs may have an advantage in modeling airway diseases such as asthma, COPD, cystic fibrosis, and many others and in studying environmental effects on lung development and homeostasis.
Divulgations
The authors have nothing to disclose.
Acknowledgements
The authors would like to acknowledge the Pluripotent Stem Cell Facility and the Confocal Imaging Core at Cincinnati Children's Hospital. This work was supported by R21AI119236 (HJ), R21AI101375 (HJ), NIH/NCATS 8UL1TR000077-04 (HJ), U19 AI070412 (HJ) and 2U19AI70235 (GKKH).
Materials
| 15mL conical | Fisher Scientific | 14-959-49D | Protocol Step 1.1. |
| BEGM | Lonza | CC-3170 | Protocol Step 1.1. |
| Penn/Strep/Fungicide | Life Technologies | 15240-062 | Protocol Step 1.1. |
| Penn/Strep | Life Technologies | 15140-122 | Protocol Step 4.a. |
| cytosoft cytology brush | Fisher Scientific | 22-263-357 | Protocol Step 1.2. |
| trypan blue | Fisher Scientific | MT-25-900-CI | Protocol Step 2.1. |
| hemacytometer | Fisher Scientific | 02-671-54 | Protocol Step 2.1. |
| PBS | Fisher Scientific | BP2438-4 | Protocol Step 2.2. |
| Cytology Funnel Clips | Fisher Scientific | 10-357 | Protocol Step 2.2. |
| cytospin funnel | Fisher Scientific | 23-640-320 | Protocol Step 2.2. |
| Cytospin 4 | Fisher Scientific | A78300003 | Protocol Step 2.2. |
| blank slide | Fisher Scientific | S95933 | Protocol Step 2.2. |
| hema 3 stain kit | Fisher Scientific | 22-122-911 | Protocol Step 2.2. |
| Bovine Dermal Colagen, type 1 | Life Technologies | A1064401 | Protocol Step 3.2. |
| T25 flask | Fisher Scientific | 08-772-45 | Protocol Step 3.3. |
| Trypsin | Lonza | CC-5012 | Protocol Step 5.2. |
| Trypsin Neutralizing Solution | Lonza | CC-5002 | Protocol Step 5.2. |
| Fetal Bovine Serum (FBS), heat sterilized at 65°C for 30min | Sigma-Aldrich | F2442 | Protocol Step 5.5. |
| Dimethyl sulfoxide Hybri-Max™, sterile-filtered, BioReagent, suitable for hybridoma, ≥99.7% - | Sigma-Aldrich | D2650 | Protocol Step 5.5. |
| polycistonic lentivirus* | e.g. Millipore | SCR511 | Protocol Step 6.4. A commercial source of reprogramming vector is listed. We routinely use the 4-in-1 plasmid reported by Voelkel et al (PMID: 20385817) to generate VSV-G-pseudotyped polycistronic reprogramming lentivirus in-house. This plasmid can be obtained by contacting |
| polybrene | Santa Cruz Biotechnology | sc-134220 | Protocol Step 6.4. |
| Irradiated CF1 MEFs | GlobalStem | GSC-6301G | Protocol Step 6.4. |
| hESC media | See recipe included in protocol | Protocol Step 6.11. | |
| SB431542 | Stemgent | 04-0010 | Protocol Step 6.11. |
| PD0325901 | Stemgent | 04-0006 | Protocol Step 6.11. |
| Thiazovivin | Stemgent | 04-0017 | Protocol Step 6.11. |
| hESC-qualified Matrigel | BD Biosciences | 354277 | Protocol Step 6.13. |
| Corning plate, 6 well | Fisher Scientific | 08-772-1B | Protocol Step 6.13. |
| mTeSR1 | StemCell | 5850 | Protocol Step 6.13. |
| 250mL disposable filter flask (0.22µm) | Fisher | SCGP-U02-RE | |
| dispase | StemCell | 7923 | Protocol Step 7.3. |
| DMEM/F12 | Life Technologies | 11320-033 | Protocol Step 7.3. |
| cell lifter | Fisher Scientific | 08-100-240 | Protocol Step 7.4. |
| hESC Media** | Protocol Step 6.11. components should be mixed and then filter sterilized. Media can be kept at 4°C for up to two weeks. When warming media, do not leave at 37°C longer than 15 min |
||
| DMEM-F12 50/50 media | Invitrogen | 11330-032 | Final Concentration |
| KO replacement serum (KO-SR) | Invitrogen | 10828-028 | 0.2 |
| 200mM L-glutamine | Invitrogen | 25030-081 | 1mM |
| 55mM ß-mercaptoethanol | Invitrogen | 21985-023 | 0.1mM |
| 100x non-essential amino acids | Invitrogen | 11140-050 | 1x |
| 2ug/mL Basic-Fibroblast Growth Factor (b-FGF) | Invitrogen | 13256-029 | 4ng/mL |
References
- Yamanaka, S. Induced pluripotent stem cells: past, present, and future. Cell stem cell. 10 (6), 678-684 (2012).
- Boulting, G. L., et al. A functionally characterized test set of human induced pluripotent stem cells. Nature biotechnology. 29 (3), 279-286 (2011).
- Thomson, J. A., et al. Embryonic stem cell lines derived from human blastocysts. Science. 282 (5391), 1145-1147 (1998).
- Guenther, M. G., et al. Chromatin structure and gene expression programs of human embryonic and induced pluripotent stem cells. Cell stem cell. 7 (2), 249-257 (2010).
- Polo, J. M., et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol. 28 (8), 848-855 (2010).
- Ono, M., et al. Generation of induced pluripotent stem cells from human nasal epithelial cells using a Sendai virus vector. PloS one. 7 (8), e42855 (2012).
- Zhou, W., Freed, C. R. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells. 27 (11), 2667-2674 (2009).
- Brennand, K. J., et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 473 (7346), 221-225 (2011).
- Mou, H., et al. Generation of multipotent lung and airway progenitors from mouse ESCs and patient-specific cystic fibrosis iPSCs. Cell stem cell. 10 (4), 385-397 (2012).
- Stadtfeld, M., Nagaya, M., Utikal, J., Weir, G., Hochedlinger, K. Induced pluripotent stem cells generated without viral integration. Science. 322 (5903), 945-949 (2008).
- Huangfu, D., et al. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotechnol. 26 (7), 795-797 (2008).
- Kuperman, D. A., et al. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med. 8 (8), 885-889 (2002).
- Braunstahl, G. J., et al. Segmental bronchoprovocation in allergic rhinitis patients affects mast cell and basophil numbers in nasal and bronchial mucosa. American journal of respiratory and critical care medicine. 164 (5), 858-865 (2001).
- Compalati, E., et al. The link between allergic rhinitis and asthma: the united airways disease. Expert review of clinical immunology. 6 (3), 413-423 (2010).
- Crimi, E., et al. Inflammatory and mechanical factors of allergen-induced bronchoconstriction in mild asthma and rhinitis. Journal of applied physiology. 91 (3), 1029-1034 (2001).
- Djukanovic, R., et al. Bronchial mucosal manifestations of atopy: a comparison of markers of inflammation between atopic asthmatics, atopic nonasthmatics and healthy controls. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology. 5 (5), 538-544 (1992).
- Gaga, M., et al. Eosinophils are a feature of upper and lower airway pathology in non-atopic asthma, irrespective of the presence of rhinitis. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 30 (5), 663-669 (2000).
- Hurst, J. R., Wilkinson, T. M., Perera, W. R., Donaldson, G. C., Wedzicha, J. A. Relationships among bacteria, upper airway, lower airway, and systemic inflammation in COPD. Chest. 127 (4), 1219-1226 (2005).
- Gaga, M., V, A. M., Chanez, P. Upper and lower airways: similarities and differences. European respiratory monograph. 18, 1-15 (2001).
- Lopez-Guisa, J. M., et al. Airway epithelial cells from asthmatic children differentially express proremodeling factors. J Allergy Clin Immunol. 129 (4), 990-997 (2012).
- Doi, A., et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nature genetics. 41 (12), 1350-1353 (2009).
- Poole, A., et al. Dissecting childhood asthma with nasal transcriptomics distinguishes subphenotypes of disease. J Allergy Clin Immunol. , (2014).
- Ji, H., et al. Dynamic transcriptional and epigenomic reprogramming from pediatric nasal epithelial cells to induced pluripotent stem cells. J Allergy Clin Immunol. 135 (1), 236-244 (2015).
- Guajardo, J. R., et al. Altered gene expression profiles in nasal respiratory epithelium reflect stable versus acute childhood asthma. The Journal of allergy and clinical immunology. 115 (2), 243-251 (2005).
- Yamanaka, S. Patient-specific pluripotent stem cells become even more accessible. Cell Stem Cell. 7 (1), 1-2 (2010).
- Kim, K., et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nature. 29 (12), 1117-1119 (2011).
- Warlich, E., et al. Lentiviral vector design and imaging approaches to visualize the early stages of cellular reprogramming. Mol Ther. 19 (4), 782-789 (2011).
- Wicell. . Wicell Feeder Based (MEF) Pluripotent Stem Cell Protocols. , (2003).
- Harkema, J. R., Carey, S. A., Wagner, J. G. The nose revisited: a brief review of the comparative structure, function, and toxicologic pathology of the nasal epithelium. Toxicol Pathol. 34 (3), 252-269 (2006).
- Allegrucci, C., Young, L. E. Differences between human embryonic stem cell lines. Hum Reprod Update. 13 (2), 103-120 (2007).
- Yoshida, Y., Yamanaka, S. Recent stem cell advances: induced pluripotent stem cells for disease modeling and stem cell-based regeneration. Circulation. 122 (1), 80-87 (2010).
- Pasca, S. P., et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nature medicine. 17 (12), 1657-1662 (2011).
- Lai, P. S., et al. Alternate methods of nasal epithelial cell sampling for airway genomic studies. J Allergy Clin Immunol. , (2015).
- Shi, Y., et al. A combined chemical and genetic approach for the generation of induced pluripotent stem cells. Cell Stem Cell. 2 (6), 525-528 (2008).
- Okita, K., et al. A more efficient method to generate integration-free human iPS cells. Nature. 8 (5), 409-412 (2011).
- Dye, B. R., et al. In vitro generation of human pluripotent stem cell derived lung organoids. Elife. 4, (2015).
- Huang, S. X., et al. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat Biotechnol. 32 (1), 84-91 (2014).
- Wong, A. P., et al. Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nature biotechnology. 30 (9), 876-882 (2012).
- Marchetto, M. C., et al. Transcriptional signature and memory retention of human-induced pluripotent stem cells. PloS one. 4 (9), e7076 (2009).
- Nukaya, D., Minami, K., Hoshikawa, R., Yokoi, N., Seino, S. Preferential gene expression and epigenetic memory of induced pluripotent stem cells derived from mouse pancreas. Genes Cells. 20 (5), 367-381 (2015).
- Vaskova, E. A., Stekleneva, A. E., Medvedev, S. P., Zakian, S. M. ‘Epigenetic memory’ phenomenon in induced pluripotent stem cells. Acta Naturae. 5 (4), 15-21 (2013).
- Bar-Nur, O., Russ, H. A., Efrat, S., Benvenisty, N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell. 9 (1), 17-23 (2011).
- Jandial, R., Levy, M. L. Cellular alchemy: induced pluripotent stem cells retain epigenetic memory. World Neurosurg. 75 (1), 5-6 (2011).
- Kim, K., et al. Epigenetic memory in induced pluripotent stem cells. Nature. 467 (7313), 285-290 (2010).
- Ohi, Y., et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nat Cell Biol. 13 (5), 541-549 (2011).
