Smart materials have attracted significant attention due to their potential for reversible "on-demand" responses to external and/or environmental signals. Temperature-responsive materials have attracted particular interest due to their lower critical solution temperature (LCST) behavior, resulting in temperature-driven precipitation at temperatures T>LCST1,2. In the context of thermoresponsive hydrogels, this lower critical solution temperature behavior is manifested by reversible swelling/de-swelling events that result in temperature-tunable bulk sizes (larger at T<LCST)3, pore sizes (larger at T<LCST)4, and interfacial properties (more hydrophilic at T<LCST)5. Such transitions have been widely applied in drug delivery (for external or environmentally-triggerable drug release4,6,7), tissue engineering and cell culture (for thermoreversible cell adhesion/delamination8,9,10), separations (for switchable membrane porosities and permeabilities or thermally-recyclable diagnostic supports11,12,13), microfluidic processes (for on-off valves regulating flow14,15), and rheological modifiers (for temperature-tunable viscosities16). The most commonly investigated thermoresponsive hydrogels are based on poly(N-isopropylacrylamide) (PNIPAM)17, although significant (and increasing) work has also been conducted on poly(oligoethylene glycol methacrylate) (POEGMA)2,18 and poly(vinylcaprolactam) (PVCL)19,20. POEGMA has attracted particular recent interest given its anticipated improved biocompatibility21,22and its facile-to-tune LCST behavior, in which linearly-predictable mixtures of monomers with different numbers of ethylene oxide repeat units in their side chains can alter the LCST from ~20 °C to >90 °C2,23. However, each of these polymers is prepared by free radical polymerization and thus contains a carbon-carbon backbone, significantly limiting the potential utility and translatability of such polymers in the context of biomedical applications in which degradation (or at least the capacity for clearance through renal filtration) is typically a requirement.
In response to this limitation, we have recently reported extensively on the application of hydrazone chemistry (i.e., the reaction between hydrazide and aldehyde-functionalized pre-polymers) to prepare degradable analogues of thermoresponsive hydrogels24,25,26,27,28,29. The rapid and reversible reaction between hydrazide and aldehyde groups upon mixing of the functionalized precursor polymers30 enables both in situ gelation (enabling facile injection of these materials without the need for surgical implantation or any type of external polymerization stimulus such as UV irradiation or chemical initiation) as well as hydrolytic degradation of the network at a rate controlled by the chemistry and density of the crosslinking sites. Furthermore, by maintaining the molecular weight of the pre-polymers used to prepare the hydrogels below the renal filtration limit, hydrogels made using this approach degrade back into the oligomeric precursor polymers that can be cleared from the body25,27,28. Coupled with the low cytotoxicity and low inflammatory tissue response induced by these materials25,26,27, this approach offers a potentially translatable method for the use of thermoresponsive smart hydrogels in medicine, particularly if well-controlled degradable analogues of such hydrogels on all length scales (bulk, micro, and nano) can be fabricated.
In this protocol, we describe methods for making synthetic thermoresponsive pre-polymers functionalized with controlled numbers of hydrazide and aldehyde groups as well as methods to apply these polymers to create hydrogels with well-defined dimensions on various length scales. In particular, this manuscript describes four distinct approaches we have developed to control the mixing of the reactive hydrazide and aldehyde-functionalized pre-polymers and thus create thermoresponsive hydrogel networks with well-defined geometries and morphologies:
To create degradable bulk hydrogels with defined sizes, a templating strategy is described by which the reactive pre-polymers are loaded into separate barrels of a double-barrel syringe equipped at its outlet with a static mixer and subsequently co-extruded into a silicone mold with the desired hydrogel shape and dimensions21,27 (Figure 1).
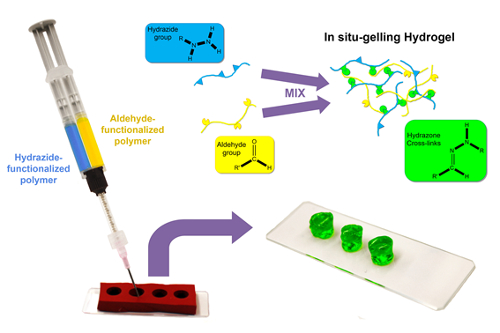
Figure 1: Schematic of bulk hydrogel formation. Hydrazide and aldehyde-functionalized polymer solutions (in water or aqueous buffer) are loaded into separate barrels of a double barrel syringe and then co-extruded through a static mixer into a cylindrical silicone mold. Rapid in situ gelation upon mixing forms a hydrazone crosslinked hydrogel, which is free standing (once the mold is removed) within seconds to minutes depending on concentration and functional group density of the precursor polymers. Please click here to view a larger version of this figure.
To create degradable gel particles on the micron-scale, a reactive microfluidics method is described in which precursor polymer solutions are simultaneously mixed and emulsified using a soft lithography-templated microfluidic chip design, enabling the formation of mixed reactive polymer droplets that subsequently gel in situ to form gel microparticles with sizes templated by the emulsion (Figure 2)31,32.
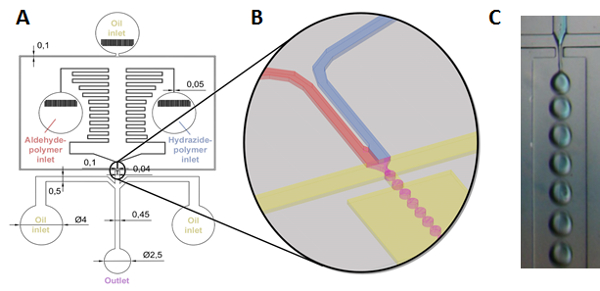
Figure 2: Schematic of gel microparticle formation via reactive microfluidics. (A,B) Hydrazide and aldehyde-functionalized polymer solutions (in water or aqueous buffer) are fed by syringe pump into separate reservoirs that are connected downstream across a zig-zag series of channels designed to create a pressure gradient preventing backflow. The polymers are then mixed just before being sheared by paraffin oil flowing from both sides (also driven by a syringe pump) and forced through a nozzle, resulting in flow-focusing production of aqueous (polymer solution) droplets in a continuous paraffin oil phase (see (B) for an illustration of the nozzle area and the droplet formation process). An additional two paraffin oil inlets are positioned after the nozzle to further separate the droplets in the collection channel to allow for complete gelation prior to particle removal from laminar flow, after which the resulting microparticulate gels are collected in a magnetically stirred beaker; (C) Picture of droplet generation process at the nozzle (note that hydrazide polymer is labeled as blue to illustrate mixing)
To create degradable gel particles on the nanoscale, a thermally-driven reactive self-assembly method is described in which a solution of one of the reactive precursor polymers (the "seed" polymer) is heated above its LCST to form a stable nanoaggregate that is subsequently crosslinked by the addition of the complementary reactive precursor polymer (the "crosslinking" polymer); the resulting hydrazone crosslinked nanogel has a size templated directly by the nanoaggregate (Figure 3)28.
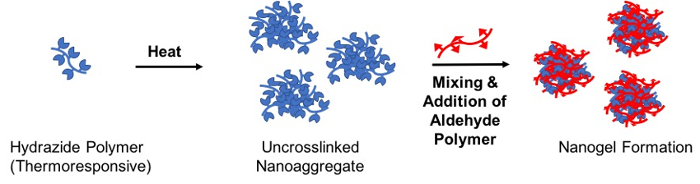
Figure 3: Schematic of nanogel formation via thermally-driven reactive self-assembly. An aqueous solution containing the (thermoresponsive) hydrazide-functionalized polymer is heated above its lower critical solution temperature to create a stable uncrosslinked nanoaggregate. Following, an aldehyde-functionalized polymer is added to crosslink the nanoaggregate via hydrazone bond formation and thus stabilize the nanogel particle upon cooling below the LCST. Please click here to view a larger version of this figure.
To create degradable nanofibers, a reactive electrospinning technique is described in which a double barrel syringe equipped with a static mixer at its outlet (as used for making bulk hydrogels) is attached to a standard electrospinning platform (Figure 4)33.
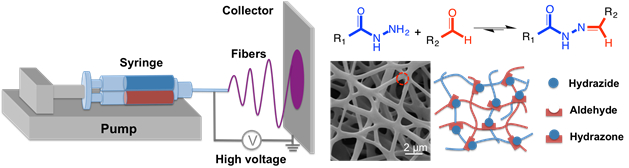
Figure 4: Schematic of hydrogel nanofiber formation via reactive electrospinning. A double barrel syringe with a static mixer (loaded as described for bulk hydrogels but also including a fraction of high molecular weight poly(ethylene oxide) as an electrospinning aid) is mounted on a syringe pump, with the needle at the end of the syringe connected to a high voltage power supply. Hydrazone crosslinking occurs during the fiber spinning process so that when the stream hits the collector (either aluminum foil or a rotating aluminum disk) the nanofibrous morphology is maintained. Please click here to view a larger version of this figure.
The application of such methods for creating degradable smart hydrogel networks is demonstrated in this protocol using either PNIPAM or POEGMA as the polymer of interest; however, the basic approaches described can be translated to any water-soluble polymer, albeit with suitable adjustments for viscosity and (in the case of the self-assembly nanogel fabrication method) the stability of the pre-polymer in forming the seed nanoaggregate.
Bulk hydrogels extruded from a double barrel syringe into a silicone mold conform to the dimensions of the mold and become free-standing upon mold removal; gelation typically occurs seconds to minutes following co-extrusion depending on polymer precursors used. Typical characterization via swelling (measured gravimetrically using a cell culture insert to easily remove the hydrogel from the swelling solution), thermoresponsivity (measured using the same technique but cycling the incubation temperature above and below the phase transition temperature), degradation (measured using the same technique but over longer time periods), and shear or compressive modulus (measured using 2 mm thick and 7 mm diameter molded samples) demonstrates the tunability of the hydrogel responses depending on the chemistry of the precursor polymer (specifically, for POEGMA, the ratio of short to long chain OEGMA monomers used to prepare the hydrogel), the mole fraction of functional groups on the precursor polymers, and the concentration of those precursor polymers (Figure 5)27.
Microfluidics leads to the formation of well-defined gel microparticles on the size scale of 25-100 µm, with the size controllable based on the flow rates of the oil and/or the combined aqueous polymer phases (Figure 6A)31. Hot stage optical microscopy confirms that the gel microparticles maintain the thermoresponsive nature of the bulk hydrogels, showing reversible temperature-dependent swelling-deswelling with only a slight hysteresis on cycle 1 (attributable to irreversible hydrogen bond formation between neighboring amide groups in the collapsed state34) consistent with that observed in bulk PNIPAM hydrogels (Figure 6B)32. Furthermore, the gel microparticles degrade back to their oligomeric precursors over time, enabling renal clearance (Figure 6C)32.
Self-assembly driven by the nanoaggregation of a hydrazide-functionalized PNIPAM polymer in a heated solution followed by crosslinking with an aldehyde-functionalized PNIPAM polymer results in highly monodisperse nanogels (polydispersity <0.1) on the size range of 180-300 nm, depending on the process conditions used (Figure 7A)28. The nanogels retain the typical thermoresponsive behavior of conventional free-radical crosslinked PNIPAM nanogels, with lower degrees of thermal deswelling observed as more cross-linking polymer was added (Figure 7B). The nanogels can be lyophilized and redispersed without a change in particle size (Figure 7C) and degrade over time via hydrolysis to re-form the oligomeric precursor polymers used to formulate the nanogels (Figure 7D).
Reactive electrospinning creates a nanofibrous hydrogel structure (Figure 8A), with nanofiber diameters on the order of ~300 nm achievable without visible electrosprayed particles present33. Soaking the POEGMA-based nanofibers in water results in rapid hydration (roughly two orders of magnitude faster than achieved with a bulk gel of the same composition, Figure 8B) but maintains the nanofibrous morphology over 8-10 weeks prior to hydrolytic degradation at physiological conditions; faster degradation is observed in acid-catalyzed environments, as expected due to the potential for acid-catalyzed hydrazone bond degradation (Figure 8C). The nanofibrous structures are also mechanically robust in both the dry and swollen states over multiple cycles, enabling easy handling and repetitive straining (Figure 8D).
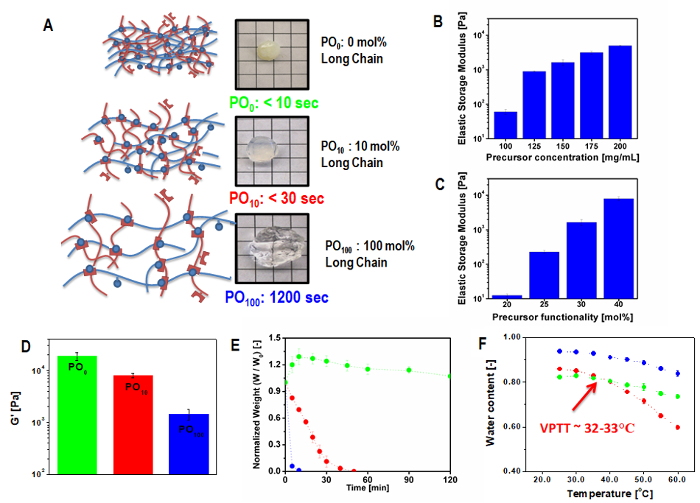
Figure 5: Properties of in situ-gelling bulk degradable thermoresponsive hydrogels. (A) Representative POEGMA gel network microstructures and bulk hydrogel images with corresponding gelation times as a function of the mole % incorporation of OEGMA475 in the precursor polymers; (B-C) Storage modulus of PO100 hydrogels by varying (B) precursor polymer concentration and (C) mole % functional group incorporation per precursor polymer; (D-F) Physiochemical properties of POEGMA hydrogels as a function of OEGMA475 mole % incorporation: (D) storage modulus (E) degradation profile in 1 M HCl, and (F) volume phase transition temperature in response to temperature change over the range 20-60 °C. All error bars represent the standard deviation of n=4 replicate measurements. Adapted from reference27 with permission from Elsevier. Please click here to view a larger version of this figure.

Figure 6: Properties of degradable gel microparticles from reactive microfluidics. (A) Effect of paraffin oil flow rate on (purified) gel microparticle size in water; (B) Thermoresponsivity of purified gel microparticles in water following a single thermal cycle above and below the volume phase transition temperature; (C) Visual assessment (photos) and gel permeation chromatography traces (graph) confirming degradation of gel microparticles back to their precursor polymer components (here, in 1 M HCl to facilitate accelerated degradation on the time scale of imaging); scale bar = 100 µm. Adapted from reference32. Please click here to view a larger version of this figure.
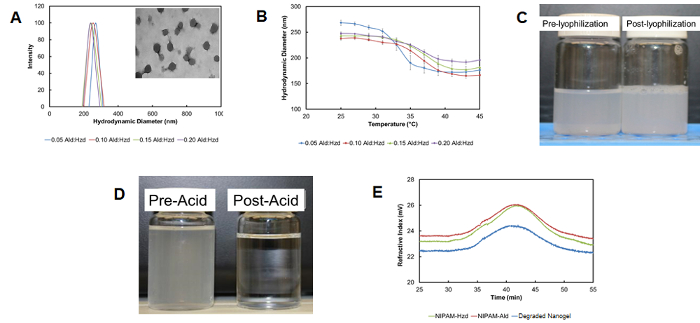
Figure 7: Properties of degradable nanogels from reactive self-assembly. (A) Particle size distributions of nanogels prepared with different aldehyde:hydrazide polymer mass ratios from dynamic light scattering (inset: transmission electron micrograph confirming the spherical nature of the nanogels); (B) Thermosensitivity of self-assembled particles as a function of the mass ratio between aldehyde:hydrazide polymer used to prepare the nanogels (from dynamic light scattering), with error bars representing the standard deviation of n=4 replicates; (C) Visual confirmation of the lack of nanogel aggregation both pre and post-lyophilization; (D) Visual confirmation of the acid-catalyzed degradation of nanogels (here in 1 M HCl for consistency with other studies above); (E) Gel permeation chromatograph traces of nanogel degradation products indicating their similarity to the hydrazide and aldehyde-functionalized precursor polymers. Adapted with permission from reference28. Copyright 2015, American Chemical Society. Please click here to view a larger version of this figure.
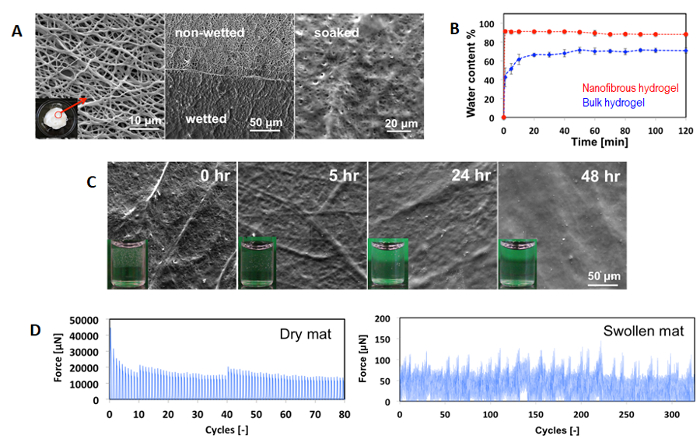
Figure 8: Properties of degradable nanofibers from reactive electrospinning. (A) Scanning electron microscopy images of nanofibers in the dry state (left), half dipped in water (middle, thin film), and fully soaked in water overnight (right, thick scaffold); (B) Swelling of nanofibrous hydrogel (red) relative to a bulk hydrogel (blue) of the same composition, with error bars representing the standard deviation of n=4 replicates; (C) Scanning electron microscopy and (inset) visual images tracking the acid-catalyzed degradation of nanofibers in 1M HCl; (D) Tensile cycling of dry (80 cycles, 20% elongation/cycle) and swollen (325 cycles, 10% elongation/cycle in 10 mM PBS) electrospun nanofibrous hydrogels. Figure modified from reference33 and reproduced with permission from the Royal Society of Chemistry. Please click here to view a larger version of this figure.
