All experiments were performed under project license number 6004219 in strict adherence to the Animals (Scientific Procedures) Act of 1986 and the UK Home Office Codes of Practice for the use of animals in scientific procedures.
1. PSM Explant Dissection
- Obtain the tail tissue from embryos produced by the timed mating of wild-type (CD1) mice13. Briefly, at embryonic day (E) 10.5, euthanize the pregnant donor mouse in a carbon dioxide chamber. Harvest the uterine horn and place it into 1x sterile phosphate-buffered saline (PBS) solution in accordance with Home Office License procedures or equivalent local rules. Transfer the uterine horn to a tissue culture dish containing fresh, sterile PBS. Perform all subsequent dissection steps in this solution.
- Under a stereomicroscope, cut the thick muscular membrane of the uterine horn using curved scissors and extract each embryo carefully using fine forceps. Take care to ensure that the tail tissue is not damaged in this process. Using curved scissors and fine forceps, dissect away the amniotic sac from each embryo, taking care not to damage the embryo.
- Use either a surgical needle or curved scissors to harvest the tail tissue of each embryo by cutting the embryo posterior to the rear limb buds.
- Balance the tail tissue ventral-side down using both forceps and a needle. Generate pairs of PSM explants from each embryonic tail by dissecting the tail tissue into two halves along the midline; perform a gentle rocking motion with a needle. Ensure that the neural tube, notochord, and PSM tissue are equally divided between the two explants.
- Pipette each contralateral PSM explant onto the underside of a 35-mm plastic culture dish lid in a small volume of pre-warmed (37 °C) culture medium (DMEM-F12 + 0.1% L-glutamine substitute supplemented with 10% fetal calf serum, 10 nM human Bfgf, and 1% penicillin/streptomycin).
- Place the dish on the top of the lid and quickly invert it so that the PSM tissue is suspended from the lid in a "hanging drop" of medium. Culture the PSM explants in a humidified chamber at 37 °C for 1 – 2 h.
- Transfer pairs of PSM explants to the individual wells of a 24-well tissue culture plate. Incubate in 4% paraformaldehyde in PBS for 1 h at room temperature (RT) or 4 °C overnight (O/N). CAUTION: Paraformaldehyde is toxic, and appropriate safety measures must be taken when working with this solution.
NOTE: Perform all subsequent washing and incubation steps in a 24-well tissue culture plate. - Wash the sample wells in PBS at RT on a rocking platform, using a fine plastic Pasteur pipette to exchange the PBS solution on the samples for fresh PBS 3 – 4 times. Process one PSM explant from each pair using immunohistochemistry (step 2) and the other using fluorescent in situ hybridization for a known clock gene (step 3).
2. Immunohistochemistry of PSM Explants
- Wash one PSM explant from each embryonic pair generated in step 1 in 2% Triton X-100 in PBS for 1 h at RT on a rocking platform, and then rinse samples briefly in PBS. Replace the PBS on the samples with blocking solution (2% bovine serum albumin (BSA) and 10% normal goat serum (NGS) in PBS + 0.1% Tween-20) and incubate O/N at 4 °C on a rocking platform.
NOTE: All subsequent washes and incubation steps in this section must be performed at RT on a rocking platform, unless otherwise stated. Wash solutions can be easily changed using a fine-tipped plastic or glass Pasteur pipette. - Dilute the desired primary antibody/antibodies in working buffer (0.1% BSA, 0.3% NGS, and 0.2% Triton X-100 in PBS). In this example, dilute the Dll1 and Notch1 antibodies 1:25 in working buffer.
NOTE: Optimization will be required to determine the appropriate dilution factor required in this step if alternative antibodies are used. - Incubate explants in the antibody solution for 3 – 5 days at 4 °C on a rocking platform. Be sure to include some samples with working buffer containing no primary antibody to act as secondary antibody controls.
- Recover the primary antibody solution in a 1.5-mL storage tube using a pipette and store it at 4 °C.
NOTE: Recovered primary antibody can be used several times, depending on the antibody used. - Perform 2 washes of the samples for 5 – 10 min each in PBS, followed by 3 washes for 10 min each in 2% Triton X-100 in PBS at RT on a rocking platform.
- Dilute fluorescently labeled secondary antibody/antibodies (epitope-matched to the primary antibody/antibodies used) in working buffer. Optionally, add 20 µg/mL Hoechst 33342 to this solution to counterstain the nuclei.
NOTE: Optimization may be required to determine the appropriate dilution factor required in this step. In this example, a dilution factor of 1:400 was typically used. - Centrifuge the secondary antibody solution for 10 min at 16 x g to prevent the formation of antibody aggregates. Add 250 – 500 µL of the secondary antibody solution to each sample well, taking care not to use the last few microliters of the solution, which may contain antibody aggregates.
- Cover the sample plate with tin foil to minimize light exposure and incubate the samples in the secondary antibody solution for 3 – 5 days at 4 °C in the dark.
- Prior to sample mounting, wash the samples twice for 10 min each in 0.1% Tween-20 in PBS (PBST) and once for 5 min in PBS at RT on a rocking platform (see step 4).
3. Fluorescent In Situ Hybridization (FISH) of PSM Explants
- If stored in an alternative vessel, transfer the remaining contralateral PSM explants to the individual wells of a 24-well tissue culture plate.
- Wash the samples for 10 min in 50% ethanol in PBST, and then perform 2 washes for 10 min each in 100% ethanol on a rocking platform at RT to dehydrate the tissue.
NOTE: All subsequent washes and incubation steps in this section must be performed at RT on a rocking platform, unless otherwise stated. - Rehydrate the tissue by washing for 10 min in 50% ethanol in PBST, followed by washing twice for 5 min each in PBST.
NOTE: Steps 3.2 and 3.3 are necessary fixation steps required for this protocol and cannot be omitted. - Incubate the samples with 10 µg/mL proteinase K in 0.1% Tween-20 in PBS (PBST) for 5 min without agitation. Quickly remove the proteinase K and rinse the samples briefly with PBST before post-fixing the tissue for 30 min in 4% formaldehyde + 0.1% glutaraldehyde in PBST. CAUTION: Both formaldehyde and glutaraldehyde are toxic, and appropriate safety measures must be taken when working with these solutions.
NOTE: The following washing and incubation steps involving 50% and 100% hybridization mixes (steps 3.6 – 3.9) should be performed without agitation. - After washing the samples twice for 10 min each in PBST, wash the samples once in 50% hybridization mix (suitable for intronic probes: 50% formamide, 5x saline-sodium citrate (SSC), 5 mM EDTA, 50 µg/mL tRNA, 0.2% Tween-20, 0.1% SDS, and 100 µg/mL heparin) in PBST prepared at RT. Incubate the samples in this solution for 10 min at 65 °C without agitation.
- Rinse the samples twice with pre-warmed (65 °C) hybridization mix before incubating the samples in hybridization mix for ≥ 2 h (up to 48 h) at 65 °C (longer incubation times improve the resulting signal-to-noise contrast). Remove the hybridization mix from the previous step, and replace it with 0.25 – 0.5 mL of pre-warmed (65 °C) hybridization mix containing a digoxigenin (DIG)-labeled anti-sense RNA probe against a known segmentation clock component.
NOTE: For example, an intronic Lunatic fringe (Lfng(i)) probe was used at a concentration of 20 µL/mL to detect nascent Lfng mRNA. The dilution used in this step is probe-dependent and will require optimization. - Seal the plate using sticky tape to prevent evaporation and incubate the samples in the probe solution for two nights at 65 °C.
- Using a fine-tipped plastic Pasteur pipette, recover the probe for reuse and store it at 20 °C. Rinse the samples twice with pre-warmed (65 °C) post-hybridization mix (50% formamide, 0.2% Tween-20, and 1x SSC) before washing the samples two more times for 20 min each at 65 °C in pre-warmed post-hybridization mix.
- Wash the samples for 15 min at 65 °C in pre-warmed 50% hybridization mix in 0.1% Tween-20 in Tris-buffered saline solution (TBST). Rinse the samples twice with TBST before washing for 30 min at RT in TBST on a rocking platform.
- Pre-incubate the explants in blocking solution (TBST + 2% blocking buffer reagent (BBR) + 20% heat-treated goat serum) for a minimum of 2 h. Replace this solution with fresh blocking solution containing a 1:200 dilution of horseradish peroxidase (HRP)-conjugated anti-digoxigenin antibody. Incubate the samples O/N at 4 °C.
- After the antibody incubation, rinse the samples 3 times with TBST at RT and transfer them to individual wells of a new 24-well tissue culture plate. Wash the explants with TBST 3 times for 1 h each.
- At this point, transfer the samples into 0.5-mL storage tubes or the individual wells of a 48-well tissue culture plate to reduce the required volume of the Tyramide signal amplification (TSA) detection reagents in the following steps.
- Incubate samples in TSA amplification buffer (see the reagents list) at RT for 1 min without agitation using as small a volume as possible, ensuring that the samples are fully immersed in the solution.
- Add TSA reagent (see the reagents list) to the sample amplification buffer at a dilution of 1:50. Quickly mix the solution until the TSA reagent is evenly distributed, cover the plate or tubes in tin foil, and incubate the samples for 60 – 90 min in the dark.
- Remove the TSA amplification solution and wash the samples in TBST 3 times for 5 min each. Transfer the explants back to a 24-well tissue culture plate to increase the wash volume and incubate the samples in 1% hydrogen peroxide in TBST for 1 h. Wash the samples with TBST 3 times for 5 min each, and then twice for 5 min each with PBST prior to sample mounting (see step 4).
4. Sample Preparation for Imaging
- Prepare one charged adhesion glass slide for each explant pair by adding 0.12-mm thick imaging spacers, which prevent the samples from being crushed by the addition of a coverslip. Remove the adhesive liner from one surface of a spacer and place it adhesive-side down onto a glass slide, pressing firmly to seal the spacer to the slide.
NOTE: For the remaining steps, endeavor to keep the samples in low light or in darkness to avoid photobleaching. Pipette explant pairs onto a prepared slide using a glass Pasteur pipette within the center of the spacer, ensuring that the dissected side of the explant faces the slide. Arrange contralateral pairs of explants side by side. - Remove as much liquid as possible from the slide using a glass Pasteur pipette and wick off any residual moisture surrounding the samples using a piece of folded low-lint tissue paper.
- Allow the samples to adhere to the slide for 45 – 60 s, until the tissue begins to appear sticky and translucent. During this time, remove the remaining adhesive liner from the spacer using forceps. Do not allow the samples to dry out.
- Add a large drop of dual-function mountant and clearing solution (0.5% p-phenylenediamine and 20 mM Tris, pH 8.8, in 90% glycerol) to the samples within the center of the spacer. NOTE: This solution turns brown/black when permitted to oxidize.
- Carefully place a circular coverslip (no. 1.5) across the samples, ensuring that the mountant is evenly distributed and that all edges of the coverslip make contact with the spacer. Place the cover-slipped slide upside down onto some low-lint tissue paper.
- Press down firmly to ensure that the coverslip fully adheres to the spacer and that any excess mountant is removed. Repeat until no more mountant blots the paper.
- Clean and label the slide(s) appropriately, and store them in the dark until imaging, short-term at -20 °C or long-term at -80 °C. After removing the slides from storage, allow them to fully thaw before imaging.
- Image the mounted samples using a confocal microscope with tiled acquisition and a high magnification objective. Image the explant pairs using a 40X oil immersion objective at 4-µm z-intervals using 488-nm, 568-nm and 647-nm laser lines to excite the green, red, and far-red fluorophores, respectively, employed for protein and mRNA detection in this study12.
NOTE: Tiled images were stitched post-acquisition to form a single image for analysis.
5. Post-acquisition Image Analysis
- Use image analysis software to define a region of interest within the PSM of each experimental sample.
- To quantify expression levels, subtract background and threshold images to the level of a no-primary control sample prior to subsequent quantification. Define an origin, an axis, and a unit length for each sample.
- Calculate the fluorescence intensity as a function of position along the normalized rostro-caudal axis for each of the M samples 12. After normalizing the intensity plots, place the intensity profiles side by side and obtain an intensity matrix f(i,j) that describes the intensity at the ith spatial position in the jth sample.
6. Temporal Ordering of Samples
- To infer temporal ordering of a known clock component, define its intensity matrix. Then, rearrange the columns of the intensity matrix so as to obtain a temporally periodic pattern. To do this, define the function
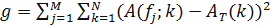
where A(fj;k) represents the autocorrelation function of the jth column of f and AT is a target autocorrelation function, chosen to enforce the temporal periodicity of the pattern, given by
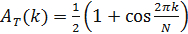
- Use the Metropolis-Hastings (or another minimization algorithm)12 to identify the order of the samples that minimize the function g. Thus, determine the order of the M samples that maximizes the temporal periodicity of a known clock component.
- Using the inferred temporal ordering of the M samples, construct an ordered kymograph for the expression pattern in the partnered channel12.
This protocol permits the visualization of the spatiotemporal profile of a protein of interest alongside clock gene transcription in the mouse PSM12. For example, Dll1 (Figure 1A-C) and Notch1 (Figure 1D-F) protein expression are shown to oscillate out of synchrony with the nascent transcription of the Notch-regulated segmentation clock gene Lfng. Quantification of Dll1, Notch1, and Lfng(i) signal intensity in relation to the antero-posterior (AP) axis of the PSM (Figure 1G) reveals clear oscillatory expression dynamics for these targets (Figure 1H-J). The spatiotemporal profile of Dll1 and Notch1 protein expression throughout the clock cycle are clearly visualized and quantified using this protocol through the post-acquisition image analysis of high-resolution fixed tissue image data.
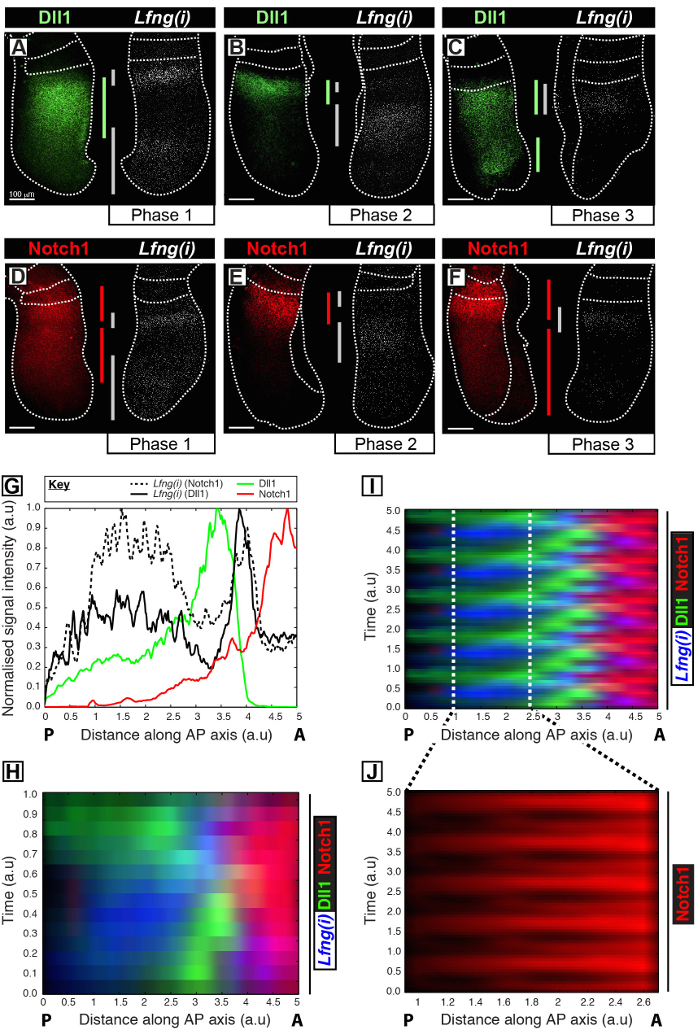
Figure 1: Spatio-temporal Visualization and Quantification of Dll1 and Notch1 Protein Expression Dynamics. (A-F) Pairs of explants from six E10.5 embryos (A-F) showing the spatial distribution of Dll1 protein (A-C) or Notch1 protein (D-F) in one half alongside the detection of Lfng pre-mRNA (Lfng(i)) in the corresponding contralateral half of each pair. Panels are arranged according to Phase 1 (A and D), Phase 2 (B and E), and Phase 3 (C and F) of the segmentation clock cycle, as determined by the spatial profile of Lfng(i) expression. The extent of the expression domains for Dll1 (green), Notch1 (red), and Lfng(i) (gray) along the antero-posterior axis of the PSM have been demarcated by color-coded bars. The dotted lines demarcate the positions of the most recently formed somite(s), the outer edges of the PSM, and the adjacent neural tissue (C and E). Scale bars (bottom left of each panel, A-F) represent 100 µm. (G) An example intensity plot depicting the axial variation in signal intensity across the PSM. The data is plotted from two explant pairs showing Lfng pre-mRNA (black hashed line) in one explant compared to Notch1 protein (red) in the contralateral explant (Embryo 1), as well as Lfng pre-mRNA (black solid line) in another explant compared to Dll1 protein (green) in the contralateral explant (Embryo 2). Measured signal intensity (y-axis) is plotted against axial position (x-axis; anterior PSM [A] to the right and posterior PSM [P] to the left). (H) A kymograph showing the spatial distribution of Dll1, Notch1, and Lfng(i) across numerous PSMs. Each row of the kymograph represents the signal intensity of an individual PSM explant. Rows are arranged in temporal sequence according to the spatiotemporal distribution of Lfng pre-mRNA (I) The spatiotemporal distribution of Dll1, Notch1, and Lfng(i) through multiple clock oscillations is simulated by the periodic extension of the data shown in (H), highlighting the oscillatory nature of Dll1 and Notch1 expression dynamics. (J) Pulsatile Notch1 protein expression in the caudal PSM is highlighted by magnification of the region demarcated in the virtual kymograph shown in (I). Modified from Reference 12. Please click here to view a larger version of this figure.
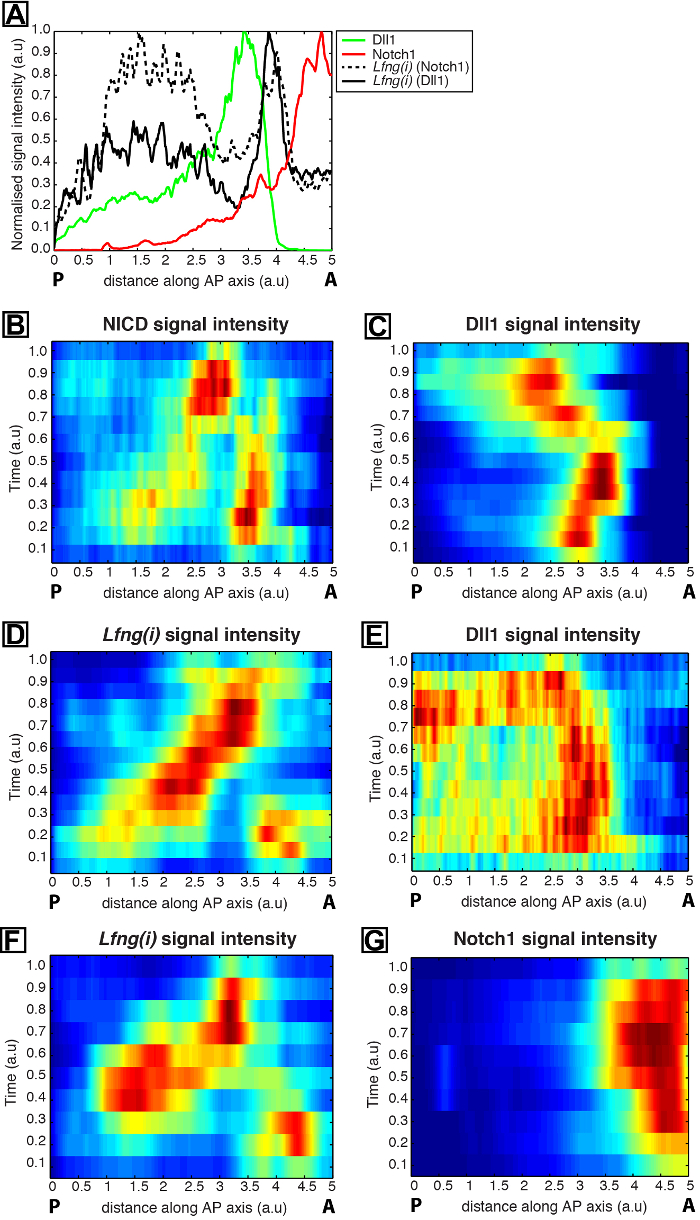
Figure 2: Quantification of the Spatio-temporal Dynamics of Dll1 and Notch1 Protein Expression. (A) An example intensity plot depicts axial variation in signal intensity across the PSM. Data plotted from two explant pairs showing Lfng pre-mRNA (black hashed line) in one explant compared to Notch1 protein (red) in the contralateral explant half, as well as Lfng pre-mRNA (black solid line) in a half explant from a second tail compared to Dll1 protein (green) in the contralateral explant half of the second tail. Measured intensities (y-axis) are plotted against axial position (x-axis; rostral [A] to the right and caudal [P] to the left). (B-H) Kymographs show the spatial distribution of Notch1, Dll1, NICD, and Lfng(i) across numerous PSMs. (B and C) NICD (B) and Dll1 (C) expression in PSM sections; (D and E) Lfng(i) (D) and Dll1 (E) in contralateral explant halves; (F and G) Lfng(i) (F) and Notch1 (G) in contralateral explant halves. From Reference 12. Please click here to view a larger version of this figure.
