Over 200 unnatural amino acids of various chemical and physical properties have been genetically incorporated into proteins in E. coli, yeast and mammalian cells3. The unnatural amino acid is incorporated in response to a specific stop codon via an orthogonal engineered tRNA/synthetase pair. The genetical approach to modify proteins has provided valuable insights into protein structure and function. Here, we present a protocol for using Voltage-Clamp Fluorometry (VCF) in combination with a fluorescent UAA.
In VCF, the simultaneous observation of functional data and structural rearrangements localized around the fluorescent probe (~5 Å) allows us to obtain dynamic information with millisecond resolution1. The fluorescent probes alter their quenching state upon localized movement of the protein. A movement of only 1-2 Å is sufficient to lead to significant changes in the fluorescence intensity4. After identification of the site of interest in the target protein, the site is mutated by point mutation. Classically, the residue had been mutated to a cysteine whereas now, an amber stop codon (TAG) is introduced for genetic fUAA incorporation. The protein is then in vitro transcribed.
While other expression systems (e.g., mammalian cells) can be used5,6,7, Xenopus oocytes are preferable for structure-function studies because of their larger size, leading to easier manipulation and higher fluorescence intensity (more fluorophores) and, therefore, to-noise ratio. Furthermore, Xenopus oocytes have low background from endogenous proteins2,8, and the dark pigmentation on the animal pole shields against background fluorescence from the cytosol. The Xenopus oocytes are surgically removed and DNA encoding the orthogonal tRNA/tRNA-synthetase pair specific for the fUAA is injected into the nucleus of the oocytes. After a 6-24 h incubation time, the protein RNA is co-injected with the fUAA into the cytosol of the oocytes, followed by a 2-3 days incubation period. In order to prevent any damage to the fUAA (photobleaching), the procedures including Anap have to be carried out under red light to avoid fluorophore excitation.
Oocytes are studied on a cut-open oocyte voltage-clamp setup, which is mounted on an upright fluorescence microscope, and electrical current and fluorescence changes are simultaneously recorded9,10. Alternatively, two-electrode voltage clamp1 or patch-clamp configurations11can be used. Fluorescence is excited by appropriate wavelengths with low RMS noise and emission recorded using a photodiode linked to an amplifier with high amplification.
There are several advantages of using fluorescent unnatural amino acids (fUAAs) in voltage-clamp fluorometry. One is access to the cytosolic side of the membrane proteins; many regulatory processes are located here (e.g., Ca2+– or nucleotide binding sites, fast and closed-state inactivation of voltage-gated ion channels, pore opening, module coupling). All these processes are now accessible for fluorescent labeling.
Another advantage is the small size of the probe leading to less disturbance of the protein. So far, two orthogonal tRNA/tRNA synthetase pairs for fUAAs have been engineered12,13, where 3-(6-acetylnaphthalen-2-ylamino)-2-aminopropanoic acid (Anap) is the only fUAA which has been used in Xenopus oocytes2,8. Anap is an environmentally sensitive fluorophore with a molecular weight of 272.3 g/mol and is only slightly bigger than tryptophan12 (Figures 1A, 1B). Due to its small size, fewer steric effects are likely to be introduced by the fluorophore compared to conventional fluorophores attached via a linker (typically more than 500 g/mol). Moreover, in the case of Anap, the fluorophore is located closer to the protein backbone than those linked to cysteines, and consequently, Anap is probing more localized rearrangements. Finally, removal of endogenous cysteines in conventional VCF in order to ensure site-specific labeling is no longer a requirement in UAA-VCF and therefore (i) leaves the proteins in (almost) their native state and (ii) allows VCF to be applied to study a wider range of proteins in which function may be altered by cysteine substitution.
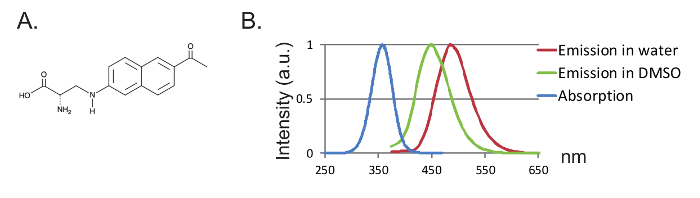
Figure 1: Anap and Fluorescence Spectra. (A) Chemical structure of Anap. (B) Normalized absorption spectrum and emission spectra for 1 nM Anap, demonstrating the sensitivity of Anap fluorescence to the solvent hydrophobicity. Emission spectra were obtained by exciting at 350 nm. Please click here to view a larger version of this figure.
A disadvantage of using fluorescent UAAs is that a heterogeneous population of proteins may result from stop codon readthrough, translational reinitiation, C-terminal truncated proteins or crosstalk with endogenous aminoacylation if the amount of aminoacylated tRNAs is scarce. Such leak expression should always be checked for in the absence of the fUAA and the tRNA/tRNA synthetase pair. We addressed the issue of translational reinitiation and how to circumvent it for N-terminal insertion sites previously14. However, when the fUAA, tRNA and tRNA synthetase are present in saturated amounts, there only remains a low probability of leak expression.
The key procedural difference between fUAA-VCF and conventional VCF is the injection and handling of the oocytes; the injection of DNA encoding the tRNA and tRNA synthetase (pAnap) is followed by the introduction of Anap, which is either co-injected with the protein mRNA or alternatively added to the incubation solution as an acetoxymethyl (AM) ester.
Frog manipulations were performed in accordance with the Canadian guidelines and have been approved by the ethics committee (CDEA, protocol #15-042) of University of Montréal.
1. mRNA Preparation for fUAA Incorporation
- Choose a site of interest in the protein where conformational changes are expected to occur. Select an amino acid in this region to be substituted for the fUAA.
NOTE: The choice of position is based on the structural rearrangements that are expected. If a high resolution structure exists and a hypothesis of the expected movements, the anap should be placed such that the chemical environment will alter; this might be either a change in the dielectric constant (hydrophobic versus hydrophilic environment) or, more likely, quenching by another amino acid. The best quenchers are tryptophans. Anap should be in contact with the quencher in one state (overlap of the van-der-Waals radii) and free of it in the other. If no high-resolution structures or models exist, one would have to scan the region of interest. In either case, it is advisable to select several nearby locations to increase the probability of obtaining expression and fluorescence signal. In order to minimize steric effects during protein maturation and/or function, one may choose to replace large and aromatic amino acids (Phe, Trp, Tyr). The authors, however, have experienced that scanning a region of interest for fUAA insertion regardless of the substituted amino acid, is more productive. - Insert an amber stop codon (TAG) at the selected site using site-directed mutagenesis15. Ensure that the protein of interest does not end on an amber stop codon (TAG). If so, mutate to a different one (ochre or opal stop codon). Amplify, isolate and sequence the DNA. Obtain protein mRNA with in vitro transcription16 and store the mRNA at 20 °C or 80 °C.
2. Oocyte Preparation and Injection
- Surgically obtain Stage V or VI oocytes from Xenopus laevis frogs and defolliculate with collagenase as previously described17.
- Anesthetize frogs with an appropriate anesthetic according to the approved animal protocol (here: 3-aminobenzoic acid ethyl ester). When they fail to respond to a gentle pinch to a toe-tip (loss of withdrawal reflex), then they are suitably anesthetized for surgery.
- Immediately remove the frogs from the anesthetic solution and thoroughly rinse its skin with fresh water. This rinse will prevent the animal from falling into deeper levels of anesthesia by removing unabsorbed chemical from the skin surface.
- Remove ovary nodes from one side surgically and carefully open the nodes using two forceps. Incubate and agitate the oocytes in "standard oocyte solution" (SOS) containing 1% (w/v) collagenase for 20-30 min to defolliculate. Wash three times with SOS solution.
- Select large and healthy oocytes individually and incubate them in Barth's solution supplemented with antibiotics (100 U/mL penicilin, 100 µg/mL streptomycin, 10 mg/100 mL kanamycin) and 5% horse serum at 18 ͦC for at least 4 h before injection.
NOTE: After 2 – 4 surgeries with a 4 month delay in between, Xenopus laevis are euthanized by prolonged (>1 h) incubation with 3-aminobenzoic acid ethyl ester.
- For nuclear injection of DNA, prepare a long and thin injection tip to be able to reach the nucleus and to avoid damaging the oocyte. Fill the injection tip with oil and mount it on the nanoinjector device.
- Install the nano-injector under a stereo microscope and use forceps to break the end of the tip. Eject oil until there is no air bubbles trapped inside the end of the tip.
- Place 1 µL of 0.1 µg/µL pAnap in nuclease-free water containing NaOH (1% of 1 N NaOH) on a piece of parafilm under a stereoscope and fill the injection tip with the DNA.
- Transfer 40 oocytes to a mesh-coated injection dish containing Barth's solution supplemented with antibiotics.
NOTE: To make the mesh-coated injection dish, cut out an appropriately sized piece of 800 µm nylon mesh to fill a polystyrene Petri dish. Add chloroform to the center and then place the mesh on top. Hold the mesh flat until the plastic sets. - As the oocyte nucleus is located in the animal (dark) pole, aim the injection tip at the center of the animal pole and impale such that the tip reaches near the center of the animal hemisphere (or 2-3x the depth compared to RNA injection). Inject 9.2 nL of pAnap into the nucleus of each oocyte. The thin tip and small injection volume may result in irregular injection or blocked tip. Occasionally check whether the injection works by injecting into the air.
NOTE: Whether the DNA gets properly injected into the nucleus is uncertain. Expect therefore 10 – 40% of the oocytes to not express the tRNA/synthetase pair. See Discussion for further elaboration.
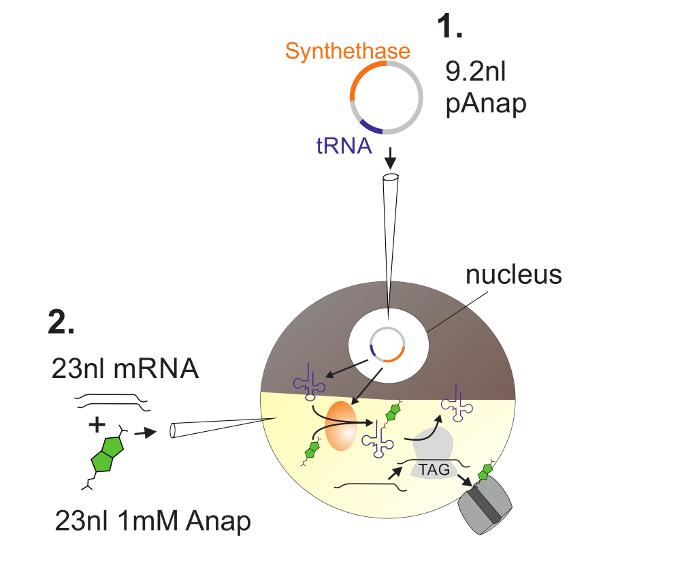
Figure 2: Illustration of DNA and RNA Injection into Xenopus Oocytes for Anap Incorporation.
First, pAnap is injected into the nucleus of the Xenopus oocyte (1). After 6-24 h, Anap and channel RNA are coinjected into the vegetal pole (2). Anap will be orthogonally aminoacylated with the tRNA bearing an amber stop anti-codon, by the aminoacyl-tRNA synthetase which is encoded by pAnap. This way, the aminoacylated Anap-tRNAs are recognized by the ribosome at the inserted amber stop codon in the channel RNA, resulting in suppression of the stop codon and insertion of Anap. Please click here to view a larger version of this figure.
- Incubate the oocytes in 2 mL Barth's solution supplemented with antibiotics and 5% Horse serum (HS) at 18 °C for 6-24 h to allow robust expression of Anap specific tRNAs and tRNA-synthetases.
NOTE: DNA incubation time can last several days before RNA injection, but it does not increase expression. - Prepare the nano-injector for RNA injection (same as step 2.2 but the injection tip does not need to be as thin as for DNA injection). Work only under red light from this point to prevent photobleaching of the Anap.
- Mix 1 µL of 1 mM Anap with 1 µL of 1-2 µg/µL mRNA directly on a piece of parafilm and fill the injection tip with the mixed solution. Impale just below the membrane in the vegetal (bright) pole and inject 46 nL in each pAnap-injected oocyte.
NOTE: The required mRNA concentration depends on the protein of interest. - Incubate the oocytes protected from light in a box or wrapped in aluminium foil, in Barth's solution supplemented antibiotics and 5% horse serum at 18 °C for 2-3 days. Exchange with fresh Barth's solution every day and remove dead oocytes to avoid contamination.
3. VCF Setup
- Install the cut-open oocyte voltage-clamp equipment as previously described18.
- Mount the electrophysiology recording system on an upright fluorescence microscope by installing the recording chamber on a slider that allows moving it between the standard stereoscope for placing the oocyte and the microscope to perform the fluorescence measurements (Figure 2c).
NOTE: The geometry of the chamber for cut-open oocytes is not suitable to use the normal transmitted light for illumination during manipulation. Therefore, a "gooseneck" halogen lamp with red filter is used to illuminate sideways from the top. The condenser of the microscope can be removed make space to lower the stage for the electrophysiology chamber. - Connect a photodiode detection system to the C-mount exit port of the fluorescence microscope (Figure 2a). Connect the photocurrent readout to a second input channel in the digital signal processor (DSP, analog/digital – digital/analog converter).
- Use a 100 W, 12 V halogen lamp as light source for the fluorescence excitation.
NOTE: Alternatively, Hg-burners may be used, but have to be reduced in intensity to prevent too rapid photobleaching during recordings. LED illumination is only recommended if the respective LEDs show significant intensity in the excitation range (e.g., ~350 nm for Anap). Most white LEDs do not reach far into the UV spectrum. - Insert an electrically-controlled shutter between excitation light source and microscope and connect its control (typically TTL-pulse) to a digital output of the DSP. Time the TTL-pulse in the recording software (see manufacturer documentation), such that the shutter opens ~100 ms before the beginning of the recording. This way, any vibration during the opening process does not interfere with the recording. The time depends on the speed and vibration of the shutter. End the pulse 5ms before the end of the recording as shown in Figure 4. This way, the value for the total fluorescence is also recorded.
- Insert an appropriate filter cube (excitation filter, dichroic mirror and emission filter) in the filter cube turret. For Anap, use Ex: 377/50 nm bandpass, dichroic 409 nm longpass, and Em: 470/40 nm bandpass.

Figure 3: VCF setup. (A) Side view of the VCF setup showing the light path inside the microscope. The filter cube contains an excitation filter, dichroic mirror and an emission filter. (B) Selected oocyte chamber dimensions are 3.4 cm for upper chamber radius (1), 5.5 cm for bottom chamber length (2), 1.4 cm for bottom chamber width (3) and 1.7 cm for middle chamber width (4). (C) Front view of the VCF setup. The first ocular on the left is for mounting the oocyte in the cut-open voltage clamp chamber and for permeabilization. Then, the chamber is slid under the microscope at the second ocular to the right. Here, the V1 electrode is inserted into the oocyte using the 4X objective, and fluorescence is recorded using the water-immersion 40X objective. Please click here to view a larger version of this figure.
4. VCF Recording
- Follow preparation steps for cut-open oocyte voltage-clamp as previously described and visualized18 (agar bridge preparation, mounting the oocyte, saponin permeabilization). However, work under red light at all time to avoid bleaching the fluorophore prior to recordings. When placing the oocyte, make sure that the animal pole faces upwards. The pigmentation under the animal pole membrane shields against autofluorescence originating from the cytosol and therefore reduces background fluorescence.
- Slide the chamber over to the microscope and focus using a 4X objective.
- Impale the oocyte with the voltage sensing V1 electrode (3 M KCl), switch to the 40X water-immersion (NA 0.8 – 0.9) objective. Focus on the animal pole which is facing upwards.
- Turn off the red light. Select the right filter cube by turning the filter cube turret and the optical exit port connected to the photodiode. Turn on the halogen lamp at highest intensity and shortly switch the shutter open for 2-5 s to read the background fluorescence intensity originating from the oocyte. With the described setup the value should be around 50-200 pA for Anap.
- Turn on the clamp, flip the bath/guard switch to active and adjust the membrane potential (V1 – V2) to the command potential by turning the knob on the I headstage.
- Select holding potential, step protocol, number and length of pulses etc. in the recording software. Record voltage-dependent currents and Anap fluorescence intensities.
5. Two-color VCF
- To monitor two locations in the same protein simultaneously, mutate an extracellular and accessible amino acid into cysteine, and remove other cysteines to ensure specific labeling with thiol-chemistry.
- Perform step 2.1-2.5.
- Prior to VCF recordings, incubate oocytes in 5 µM TMR-maleimide in labeling solution for 15 min (or other dye with non-overlapping spectra compared to Anap).
- Wash the oocytes with labeling solution three times to remove excess dye.
- Perform step 4.1-4.6.
- Insert an appropriate filter cube for TMR (excitation filter, dichroic mirror and emission filter) in the filter cube turret. Switch to the TMR filter cube by turning the filter turret.
- Read the background fluorescence for TMR as described for Anap in step 4.4.
NOTE: Labeling with thiol-chemistry results in high background fluorescence due to unspecific labeling in the membrane. Therefore, the TMR background fluorescence may saturate the amplifier (>2,000 pA). In that case, do not decrease the light intensity, but simply subtract the background fluorescence by adding an offset current to the photodiode. In commercially available systems use the "sample-and-hold" feature on the detector system. Note the background fluorescence value (using a 10X neutral density filter) in a laboratory journal, as this value will not be recorded (saturation). - Record voltage-dependent currents and TMR fluorescence intensities simultaneously as in step 4.6.
Figure 4 shows an example of VCF recordings obtained from an oocyte expressing Shaker channels with fast inactivation removed (IR), L382stop-W434F in presence of pAnap and Anap. The W434F mutation blocks the ionic potassium currents, which makes it possible to measure the transient gating charge displacements (gating currents). The simultaneous recordings of gating currents (upper trace) and Anap fluorescence intensity changes (lower trace) upon depolarization demonstrate the successful incorporation of Anap into position L382. Here, Anap is located on the bottom of each of the four S4 transmembrane helices that move during activation (L382) and thus report on intracellular local rearrangements. A fluorescence change can be caused by solvent relaxation (environmental polarity changes) and/or quenching by other amino acids4. Both mechanisms are a result of relative protein rearrangements.
Figure 5 displays Anap and TMR fluorescence signals using step protocols obtained from the same oocyte. By adding the A359C mutation into the L382stop-W434F background and labelling with TMR-maleimide, it is possible with the described technique to probe real-time movements in different regions in the same protein. In this case, TMR probes the movement of the upper S4 helix (A359C) while Anap probes the movement of the bottom of S4 (L382). From the time course of the fluorescence change one can retrieve dynamical information of the transition monitored by the fluorescence by analyzing the kinetics (Figures 5A, 5B). Moreover, the fluorescence-voltage relationship (FV) is obtained by plotting the steady-state fluorescence intensity against voltage (Figure 5C). The FV reflects the equilibrium between occupancies of monitored states which in the case of ion channels could follow voltage sensor movement, or opening of the central pore. By using fUAAs it is now possible to obtain the FV from the inside of the channel. Figure 5C shows that the upper part of S4 (TMR) has the same voltage dependency as the lower part of S4 (Anap).
The signal-to-noise ratio is mainly dependent on the relative fluorescence change (dF/F) and the total fluorescence. The dF/F defines the signal size whereas the noise is determined by the total fluorescence because of the quantum nature of light (Poisson noise). The dF/F will depend on the state of quenching in the different states of the protein, and the occupancy of the states (e.g., the open probability). The total fluorescence comprises the fluorescence from specific labeling and from unspecific background labeling. The total specific fluorescence is defined by the number of expressed proteins and the quantum yield of the fluorophore. The large surface of the Xenopus oocytes is advantageous because it increases the number of contributing proteins.
Anap's quantum yield, i.e. the number of photons emitted per excitation cycle or the "brightness" of the fluorophore, is lower, which leads to lower total fluorescence intensity and thereby lower signal-to-noise ratio. At the same time, Anap labeling leads to low background fluorescence such that the signal-to-background ratio (dF/F) is higher for Anap (Figures 5A, 5B).
An important control experiment is to check expression in absence of Anap, in order to assess the effect of leak expression. It has previously been shown that isolated voltage sensor domains (iVSDs, 1-382) which lack the S4-S5 linker and the pore are functionally expressed19. Therefore, in the absence of Anap, L382stop-W434F channels express as iVSDs as shown in Figure 6. Expression of full-length channels with Anap has little or no iVSD currents, while truncated channels exhibit strong iVSD currents in the absence of Anap. The presence of such C-terminal truncated proteins depends on the position of the inserted stop codon and should always be taken into consideration when working with unnatural amino acids. Control experiments without Anap allow verification whether a heterologous population is present in experiments with Anap.
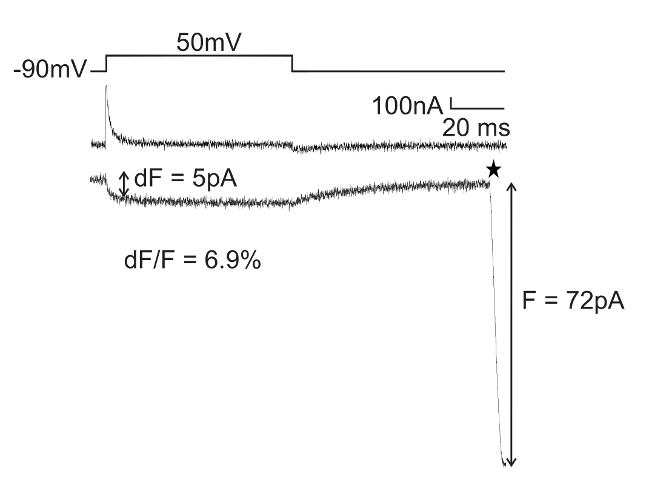
Figure 4: Simultaneous Gating Currents and Fluorescence Changes from an Cytosolic Protein Surface Obtained with VCF. Gating currents obtained with a P/4 subtraction protocol and simultaneous recording of fluorescence upon depolarization. Anap is incorporated into Shaker at position L382, giving rise to voltage-dependent fluorescence intensities from the intracellular end of the S4 helix. The maximum fluorescence intensity is denoted "F" and the fluorescence change is denoted "dF". The star at the end of the fluorescence change marks the closing of the electrical shutter (the opening of the shutter before the pulse is not shown). Please click here to view a larger version of this figure.
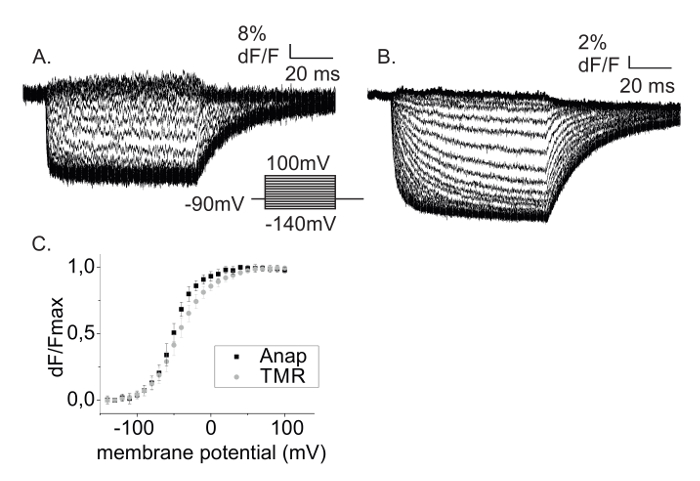
Figure 5: Two-color VCF with Anap and TMR. (A) Anap and (B) TMR fluorescence changes obtained from a Shaker A359C-L382Anap-W434F expressing oocyte labeled with TMR-maleimide. (C) Fluorescence changes from A and B are plotted against the membrane potential (FV). Data shows mean ±SD with n = 5 oocytes. Please click here to view a larger version of this figure.
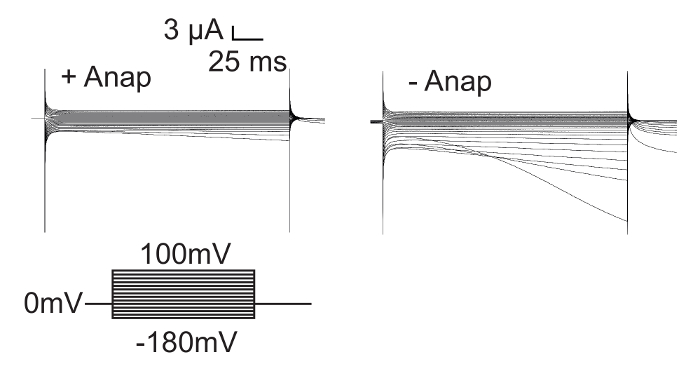
Figure 6: C-terminal Truncated Shaker Channels are Expressed in Absence of Anap. Isolated voltage sensor domains (iVSD, 1-382) are expressed with zH4IR-L382stop-W434F in the absence of Anap, resulting in iVSD currents at hyperpolarized potentials19. In the presence of Anap however, full length Shaker expression competes with iVSD expression, and as a result, the amount of iVSD is less. Please click here to view a larger version of this figure.
