Mature murine T-cells were subjected to ChIP analysis to investigate the enrichment of the histone mark mono-methylation of lysine 4 on histone 3 (H3K4me1), tri-methylation of lysine 4 on histone 3 (H3K4me3), and acetylation of lysine 27 on histone 3 (H3K27ac), as well as the nucleosome occupancy, as revealed by panH3 ChIP. The shearing quality was evaluated by analysis on a 1.8% agarose gel (Figure 2A) and on an electrophoresis system (Figure 2B). H3K4me1 and H3K4me3 are generally used to identify enhancer sites and promoters, respectively (Figure 3A). In fact, H3K4me3 is prominent but not exclusively enriched at promoters5,8,14. A characteristic of active enhancers is to be highly enriched for H3K4me1 and H3K27ac and poorly enriched for H3K4me3 (Figure 3A, upper panel), whereas inactive enhancers present low levels of H3K4me3 and H3K27ac but high levels of H3K4me1 (Figure 3A, lower panel). As shown in Figure 3B, the Glyceraldehyde 3-phosphate dehydrogenase (Gapdh) gene is representative of the analysis of active gene promoters (Gapdh promoter). In fact, it shows high levels of H3K4me3 and H3K27ac and low levels of H3K4me1. In Figure 3B, Deltex1 (Dtx1) is representative of inactive enhancers (Dtx1 enhancer), as it is highly enriched for H3K4me1 and poorly enriched for H3K4me3 and H3K27ac. Gene desert is representative of a region lacking coding genes and is usually sued as a negative control for active chromatin marks. The observation that H3K4me1, a well-accepted enhancer mark4,5,7,14,18, is not enriched at Gene desert suggests that this region lacks enhancers.
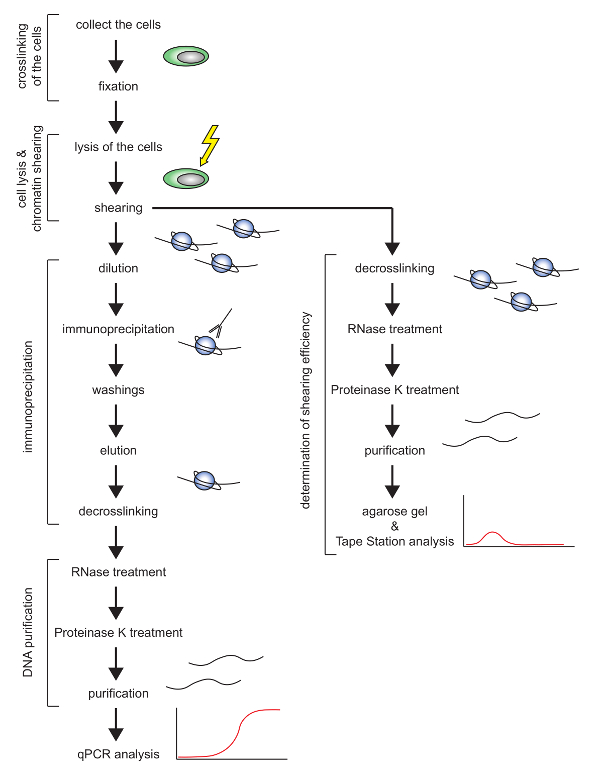
Figure 1: Schematic overview of the ChIP protocol presented. Please click here to view a larger version of this figure.
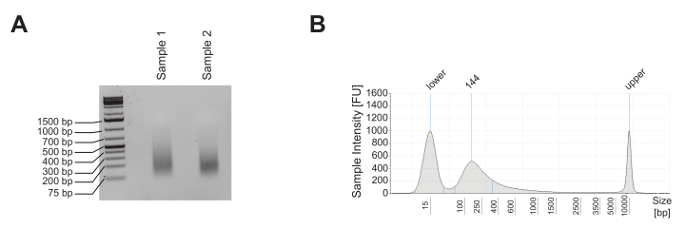
Figure 2: Shearing quality control of the mature mouse T-cell line. (A) Two different aliquots of mature mouse T-cell line E2-10HA were sheared, and approximately 500 ng of purified DNA were analyzed on a 1.8% agarose gel to evaluate their shearing quality. (B) 1 ng of purified DNA from sample 2 was analyzed by electrophoresis to evaluate its shearing quality. Please click here to view a larger version of this figure.
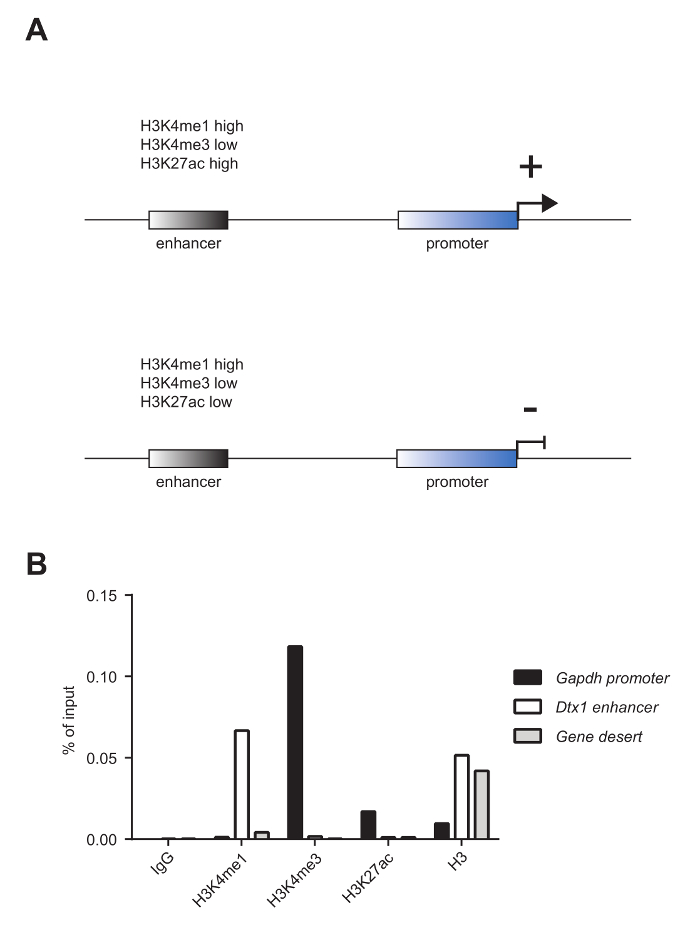
Figure 3: Chromatin marks that characterize enhancers and promoters. (A) Active enhancers (upper panel) are characterized by high H3K4me1, low H3K4me3, and high H3K27ac, whereas inactive enhancers (lower panel) present high H3K4me1, low H3K4me3, and low H3K27ac. (B) The samples presented in Figure 2A were pooled together and used for ChIP analysis versus histone marks H3K4me1, H3K4me3, and H3K27ac and histone occupancy, as revealed by panH3 ChIP. This analysis shows that the promoter of the housekeeping gene Glyceraldehyde 3-phosphate dehydrogenase (Gapdh promoter) is highly enriched for H3K4me3 and H3K27ac and lowly enriched for H3K4me1. On the other hand, the enhancer of the inactive gene Deltex1 (Dtx1 enhancer) is highly enriched for H3K4me1 but poorly enriched for H3K4me3 and H3K27ac. ChIP using a panH3 antibody was used to investigate nucleosome occupancy. Gene desert was used as a negative control. One representative experiment is shown. Please click here to view a larger version of this figure.
| Name of the buffer | Reagent | Final concentration |
| Dilution buffer | Sodium dodecyl sulfate (SDS) | 0.01% (w/v) |
| Triton X-100 | 1.1% (v/v) | |
| Ethylenediaminetetraacetic acid (EDTA) pH 8.0 | 1.2 mM | |
| Tris-HCl pH 8.1 | 16.7 mM | |
| Sodium chloride (NaCl) | 167 mM | |
| DMA solution | Dimethyl adipimidate (DMA) | 10 mM |
| Phosphate-buffered saline (PBS) | 1x | |
| Elution buffer | Sodium dodecyl sulfate (SDS) | 1% (w/v) |
| Ethylenediaminetetraacetic acid (EDTA) pH 8.0 | 10 mM | |
| Tris-HCl pH 8.0 | 50 mM | |
| High salt buffer | Sodium dodecyl sulfate (SDS) | 0.1% (w/v) |
| Triton X-100 | 1% (v/v) | |
| Ethylenediaminetetraacetic acid (EDTA) pH 8.0 | 2 mM | |
| Tris-HCl pH 8.1 | 20 mM | |
| Sodium chloride (NaCl) | 500 mM | |
| IMDM medium | Iscove's modified Dulbecco's medium (IMDM) | 1x |
| Fetal bovine serum (FBS) | 2% (v/v) | |
| Penicillin/Streptomycin | 1x | |
| Peptone primatone | 0.3 mg/mL | |
| Insulin solution human | 4.8 mg/mL | |
| Minimum essential medium non-essential amino acids (MEM NEAA) | 1x | |
| LiCl salt buffer | Lithium chloride (LiCl) | 0.25 M |
| IGEPAL-CA630 | 1% (v/v) | |
| Ethylenediaminetetraacetic acid (EDTA) pH 8.0 | 1 mM | |
| Tris-HCl pH 8.1 | 10 mM | |
| Low salt buffer | Sodium dodecyl sulfate (SDS) | 0.1% (w/v) |
| Triton X-100 | 1% (v/v) | |
| Ethylenediaminetetraacetic acid (EDTA) pH 8.0 | 2 mM | |
| Tris-HCl pH 8.1 | 20 mM | |
| Sodium chloride (NaCl) | 150 mM | |
| Protein A Sepharose beads washing buffer | Tris-HCl pH 8.0 | 20 mM |
| Sodium chloride (NaCl) | 500 mM | |
| Ethylenediaminetetraacetic acid (EDTA) pH 8.0 | 2 mM | |
| Sodium dodecyl sulfate (SDS) | 0.1% (w/v) | |
| IGEPAL-CA630 | 1% (v/v) | |
| SDS Lysis buffer | Sodium dodecyl sulfate (SDS) | 1% (w/v) |
| Ethylenediaminetetraacetic acid (EDTA) pH 8.0 | 10 mM | |
| Tris-HCl pH 8.1 | 50 mM | |
| TE buffer | Tris-HCl pH 8.0 | 10 mM |
| Ethylenediaminetetraacetic acid (EDTA) pH 8.0 | 1 mM |
Table 1: List of the buffers and the medium used in this protocol.
| ON | OFF | |
| Peak power | 150.0 | 2.5 |
| Duty factor | 15.0 | 15.0 |
| Cycles/burst | 500 | 500 |
| No. of cycles | 28 | |
Table 2: Shearing settings used in this protocol. These conditions have been optimized for a mature mouse T-cell line.
| Antibody | Supplier | Amount of antibody/immunoprecipitation | Amount of cells/immunoprecipitation | Washing conditions |
| H3 | Abcam (ab1791) | 2.5 mg | 5 x 106 | Once in low salt buffer, twice in High salt buffer, twice in LiCl salt buffer and three times in TE buffer |
| H3K4me1 | Abcam (ab8895) | 2.5 mg | 5 x 106 | Once in low salt buffer, twice in High salt buffer, twice in LiCl salt buffer and three times in TE buffer |
| H3K4me3 | Diagenode (pAb-003-050) | 2.5 mg | 5 x 106 | Once in low salt buffer, twice in High salt buffer, twice in LiCl salt buffer and three times in TE buffer |
| H3K27ac | Diagenode (pAb-174-050) | 2.5 mg | 5 x 106 | Oce in low salt buffer, twice in High salt buffer, twice in LiCl salt buffer and three times in TE buffer |
| IgG | Diagenode (C15410206) | Variable* | Variable* | Variable* |
| * In the case of the IgG control, the amount of both antibody and cells, as well as the washing steps, have to mirror the conditions of the other immunoprecipitations. | ||||
Table 3: Antibodies and washing conditions used in this study.
| Gene | forward primer | reverse primer | probe |
| Gapdh promoter (0 kb) | 5'-GGG TTC CTA TAA ATA CGG ACT GC-3' | 5'-CTG GCA CTG CAC AAG AAG A-3' | 68 |
| Dtx1 enhancer (+26 kb) | 5'-CTC TGG GTT GTA GGG GAC AG-3' | 5'-GCA TGG GAA CTG TGT TAC AGA A-3' | 27 |
| Gene desert | 5'-CAA TGC ATG GGT CCA GAT TT-3' | 5'-ATT GGC ACG GAA GTA GTG CT-3' | 94 |
Table 4: Primers and probes used for qPCR in this study.
| Problem | Causes | Solution |
| Chromatin is under sheared and fragments are too big. | Too many cells have been used. | Reduce the number of cells. |
| The shearing is too low. | Increase the number of shearing cycles. | |
| Increase the shearing peak power, the duty factor and/or the cycles/burst. | ||
| Chromatin is over sheared and fragments are too small. | Too few cells have been used. | Increase the number of cells. |
| The shearing is too high. | Reduce the number of shearing cycles. | |
| Reduce the shearing peak power, the duty factor and/or the cycles/burst. | ||
| Too low recovery over IgG control and/or negative control (high background). | Not enough chromatin used for the experiment. | Increase the amount of chromatin. |
| Amount of antibody is too low. | Increase the amount of antibody. | |
| Amount of antibody is too high. | Reduce the amount of antibody. | |
| The antibody has low specificity. | Change the antibody. | |
| Washing conditions are too stringent. | Reduce the number of washing. | |
| Reduce the stringency of the washing buffers. | ||
| Washing conditions are not stringent enough. | Increase the number of washing. | |
| Increase the stringency of the washing buffers. | ||
| Too much DNA was pipetted in the qPCR. | Reduce the amount of DNA pipetted in the qPCR. | |
| qPCR conditions are not optimal. | Optimize the qPCR conditions. | |
| Reduce the number of cycles. | ||
| No product or product is too low in the qPCR reaction of the input sample. | Not enough DNA was pipetted in the qPCR. | Increase the amount of DNA pipetted in the qPCR. |
| qPCR conditions are not optimal. | Optimize the qPCR conditions. | |
| Increase the number of cycles. | ||
| Not enough chromatin used for the experiment. | Increase the amount of chromatin. | |
| No signal in the positive control and/or panH3 ChIP. | Not enough chromatin used for the experiment. | Increase the amount of chromatin. |
| Amount of antibody is too low. | Increase the amount of antibody. | |
| The antibody has low specificity. | Change the antibody. | |
| Elution is sub-optimal. | Be sure to frequently vortex the samples to maintain the beads in solution. |
Table 5: Troubleshooting.
