The technique described here can be used to introduce any test gene construct into aleurone cells of barley grains. The level of expression from the test gene may then be conveniently measured (Figure 2). The higher throughput protocol described here greatly increases the efficiency of the seed grinding and enzyme assay steps. This method has been used to assess the ability of the transcription factor TaABF15,6 to activate expression of the ABA-induced gene HVA17. In the representative results presented here, an HVA1::GUS reporter construct was introduced into aleurone cells together with a UBI::TaABF1 effector construct (Figure 3A). While a TaABF1/HVA1 ratio of just 0.01 was sufficient to cause a 3-fold induction of the HVA1 reporter gene in the absence of exogenous ABA, higher levels of the TaABF1 construct resulted in progressively greater HVA1 expression, in a dose-dependent manner (Figure 3B,C).
This experimental protocol may also incorporate the use of hormones or other compounds during the post-bombardment incubation period to assess the role of those molecules in the regulation of gene expression. Previous work by other investigators has suggested the importance of phospholipase D in some aspects of ABA signaling. Specifically, the product of phospholipase D activity, phosphatidic acid, has been implicated as an intermediate in some responses to ABA8,9. The fact that 1-butanol, an inhibitor of phospholipase D (PLD), has been shown to prevent ABA-induced gene expression in aleurone cells suggests that PLD might be an integral part of ABA signaling in this tissue. The bombardment protocol was used to test whether 1-butanol could inhibit either ABA-induced or TaABF1-induced HVA1 expression. As expected, either exogenous ABA or TaABF1 could strongly induce expression of the HVA1::GUS reporter construct (Figure 3C). In either case, the presence of 1-butanol strongly inhibited the HVA1 induction. This suggests that PLD could play an important role in the activation of HVA1 gene expression by ABA, through TaABF1.
In addition to its role in developing and germinating grains, ABA is also critical for regulating stomatal aperture in leaves. In the guard cells of stomata, the production of phosphatidic acid is stimulated by nitric oxide (NO), which is in turn induced by hydrogen peroxide (H2O2). For some responses, NO and H2O2 can substitute for ABA9. To determine whether this is also the case in aleurone cells, the bombardment protocol was used to test whether the NO donor sodium nitroprusside (SNP) or H2O2 could substitute for ABA in inhibiting the expression of the GA-induced Amy32b gene. While ABA could strongly inhibit GA-induced expression for the Amy32b::GUS reporter construct, neither H2O2 nor SNP caused any significant reduction (Figure 3D). These results, therefore, do not support a role for NO and H2O2 in aleurone cell ABA responses.
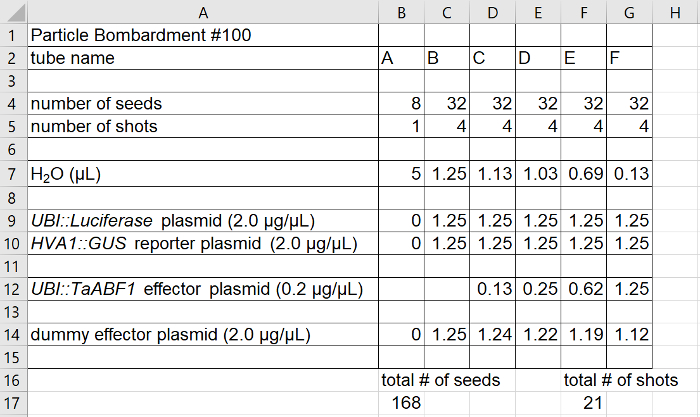
Figure 1: Spreadsheet for a typical bombardment experiment. The amount of each plasmid preparation to be placed in the tube for each bombardment is indicated. In this experiment, four shots (8 grains each) were used for each treatment to give a total of eight possible samples (4 grains each) for each treatment. In order to maintain the same total DNA concentration in each bombardment, a "dummy" effector plasmid was used to substitute for the UBI::TaBF1 effector plasmid in some cases. Treatment A is a "blank" bombardment to establish the background level of GUS and luciferase activity in aleurone cells. The results of the bombardment experiment outlined in this spreadsheet are shown in Figure 3B. Please click here to view a larger version of this figure.
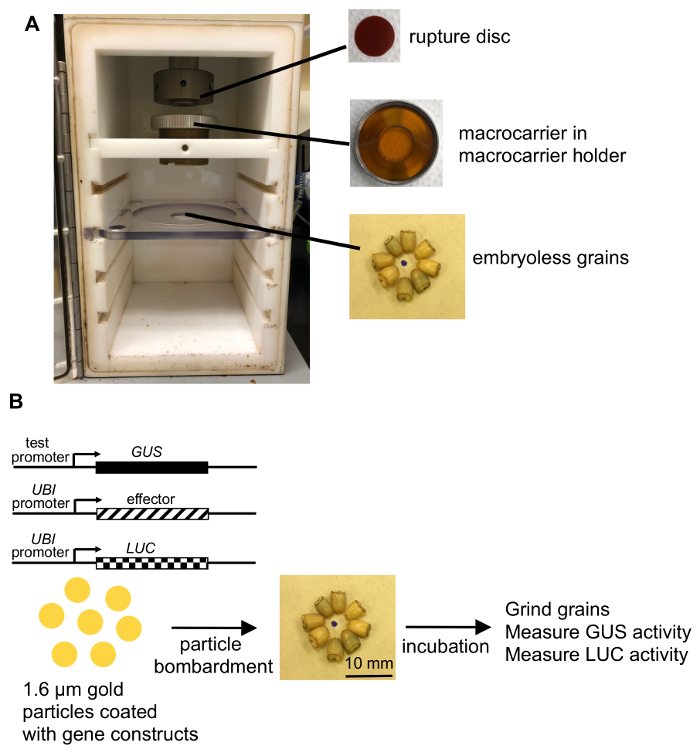
Figure 2: Diagram of the protocol. (A) Schematic of the bombardment device. (B) Gold particles coated with a reporter construct carrying the promoter to be tested, an effector (if needed), and an internal control (UBI::luciferase) are introduced into the barley aleurone cells by particle bombardment. After incubation (typically for 24 h), the grains are ground and assayed for both GUS activity and luciferase activity. Please click here to view a larger version of this figure.
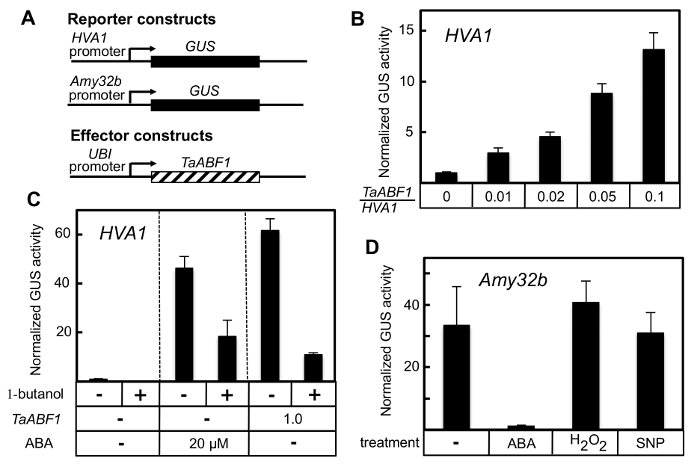
Figure 3: Regulation of HVA1 and Amy32b promoters by TaABF1, ABA, and potential mediators of ABA signaling. (A) Reporter and effector constructs used in the experiments. (B) The effector construct UBI::TaABF1 was co-bombarded into aleurone cells with the HVA1::GUS reporter and the internal control construct (UBI::luciferase). The amount of UBI::TaABF1 effector (relative to the HVA1::GUS reporter) varied from 0 to 0.1, as indicated below the bars. GUS and luciferase activities were measured after 24 h of incubation. Data shown are the means (± SE) of 6 – 8 biological replicates. (C) The HVA1::GUS reporter was co-bombarded into aleurone cells with the internal control construct (UBI::luciferase) in the presence or absence of the effector construct UBI::TaABF1. The amount of TaABF1 effector (relative to the reporter) was 1.0. GUS and luciferase activities were measured after 24 h of incubation with or without 20 µM ABA and 0.4% 1-butanol. (D) The Amy32b::GUS reporter was co-bombarded into aleurone cells with the internal control construct (UBI::luciferase). GUS and luciferase activities were measured after 24 h of incubation in the presence of 1 mM GA with or without 20 µM ABA, 1 mM H2O2, or 100 µM sodium nitroprusside (SNP). Values for GUS activity are relative to the expression of the Amy32b::GUS reporter in the absence of GA. Please click here to view a larger version of this figure.
