1. Day 1: Cell Culture
- Culture the cells to be analyzed to the desired amount. Cell culture conditions and media depend on the cell types under investigation.
NOTE: In principle, any eukaryotic cell is amenable to an analysis of chromatin accessibility by FAIRE. For this experiment, 4 x 106 HEK-293T cells are seeded in a single 10 cm dish in DMEM medium supplemented with 10% (v/v) FBS and 1% (w/v) penicillin/streptomycin, and incubated at 5% CO2 and 37 °C for 24 h until the cells reach 70 – 80% confluence (~ 8 x 106 cells per dish).
2. Day 2: Formaldehyde Crosslinking and Cell Harvesting
CAUTION: Formaldehyde is highly toxic and must be always used under a fume hood. Wear protective clothes (gloves and lab coat). Discard the waste appropriately.
- Add formaldehyde directly to the cell culture medium in a fume hood to reach a final concentration of 1% (v/v) (270 µL of 37% (v/v) formaldehyde to 10 mL of cell culture medium). Incubate for 10 min at room temperature (20 – 25 °C) and slew the plates manually every 2 min.
NOTE: The crosslinking time can be adjusted according to the cell type. For the majority of cell lines, 10 min of crosslinking is adequate. For tissues, longer incubations might be required to allow the fixation of all cells. - Add glycine to a final concentration of 0.125 M to quench the formaldehyde (add 500 µL of a 2.5 M glycine stock to 10 mL of culture medium). Incubate for 5 min at room temperature and slew every 2 min.
- Wash the cells 3x with ice cold PBS (8 g/L NaCl, 0.2 g/L KCl, 1.44 g/L Na2HPO4 · 2H2O, 0.24 g/L KH2PO4, adjust pH to 7.4 with HCl or NaOH). Aspirate the medium and wash directly in the cell culture dish or flask by adding 10 mL of PBS. Repeat this step twice being very careful in the case of loosely adherent cells such as HEK-293T. Add PBS carefully to the side of the dish in order to avoid detachment of the cells.
- After the third wash, resuspend the cell in 1 mL of ice cold PBS and transfer to a 1.5 mL reaction tube on ice. Use a cell scraper to detach the cells when necessary.
NOTE: When starting with a limited material, use low DNA-binding tubes to avoid sample loss during the procedure. - Centrifuge for 5 min at 300 x g at 4 °C in a cooled tabletop centrifuge. Discard the supernatant by carefully aspirating it.
3. Cell Lysis and Chromatin Fragmentation
- Resuspend the cell pellet in 1 mL FAIRE lysis buffer (1% (w/v), 10 mM EDTA, 50 mM Tris HCl pH 8.1, 10 µg/mL leupeptin, 10 µg/mL aprotinin, 2 mM PMSF), and resuspend the cells by carefully pipetting up and down several times. Incubate for 20 min on ice. Store the samples at -80 °C at this step if needed.
- Sonicate the crosslinked DNA to shear it to an average size of 200 – 300 bp. The specific settings of the sonicator must be determined for each source of samples and the used sonication device. In any case ensure that the DNA is efficiently sheared. An optimization step for DNA shearing is described under step 5.14. Cool the samples on ice during sonication to avoid heating of the sample.
- Centrifuge the sample for 15 min and 13,000 x g at 4 °C.
- Transfer the supernatant to a new reaction tube. Split the samples in 100 µL aliquots. Store the samples at -80 °C, if needed.
NOTE: The protocol can be interrupted at this point.
4. De-crosslinking of Control DNA
- Take a 100 µL aliquot from step 3.4 to reverse the crosslink and to be used as a reference for total DNA. Add 10 µL of RNase-A (10 mg/mL) and incubate for 1 h at 37 °C.
- Add 10 µL of Proteinase K (20 mg/mL). Use a programmable thermo-block to incubate for 4 h at 37 °C and then for 6 h at 65 °C to cleave the proteins and to reverse the crosslinks. Finally, hold the samples at 4 °C until the protocol is resumed and then proceed to step 5.
NOTE: Other FAIRE protocols use chromatin samples from cells which have not been crosslinked as reference, thus abolishing the need for de-crosslinking11. The fact that these samples are not crosslinked could lead to differences in the sonication efficiency or in DNA stability, therefore the use of total DNA reference samples that have undergone the same procedure as the test samples is more accurate.
5. Day 3: Phenol:Chloroform Purification of DNA
- Take the non-de-crosslinked aliquot from step 3.4. Add 10 µL of RNase-A (10 mg/mL) and incubate for 1 h at 37 °C.
- Take the non-de-crosslinked aliquot form step 5.1 and the de-crosslinked sample from step 4.2 and proceed in parallel. Add H2O to reach a final volume of 300 µL.
- Add 300 µL of phenol:chloroform:isoamyl alcohol (25:24:1) in a fume hood and vortex vigorously.
CAUTION: Phenol is corrosive and toxic and must be used always under a fume hood. Wear protective clothes (gloves and lab coat), make sure the reaction tubes are tightly closed, and dispose the waste appropriately. - Centrifuge for 10 min at 13,000 x g at 4 °C.
- Carefully transfer 280 µL of the upper aqueous phase to a new reaction tube. Make sure not to take any debris from the interphase which also contains proteins. Discard the remaining lower phase as organic solvent waste.
- Repeat steps 5.3 to 5.5 and carefully transfer 270 µL of the upper aqueous phase to a new reaction tube.
- Add 270 µL of chloroform in a fume hood and vortex vigorously.
NOTE: This step is used to eliminate any remaining phenol from the sample. - Centrifuge for 10 min at 13,000 x g at 4 °C.
- Carefully transfer 250 µL of the upper aqueous phase to a new reaction tube, taking care not to carry any debris from the interphase. Discard the remaining sample as organic solvent waste.
- Add 25 µL of 5 M NaCl and 250 µL of isopropanol. Add 5 µL of glycogen (20 mg/mL) as the DNA carrier. Mix well by inverting the tubes and incubate for 20 min at room temperature. Centrifuge for 10 min at 13,000 x g at 4 °C to precipitate the DNA.
- Wash the DNA pellet with 400 µL of 70% (v/v) ethanol. Centrifuge for 10 min at 13,000 x g at 4 °C.
- Aspirate the supernatant carefully and let the pellet dry for 10 min at room temperature. Resuspend in 100 µL TE buffer (10 mM Tris pH 8, 1 mM EDTA).
- Incubate the non-de-crosslinked free DNA sample for 4 h at 65 °C to remove inter-DNA crosslinks. Store the samples at -20 °C.
- Quantify the amount of DNA using a spectrophotometer and run an aliquot to check the fragment size on a 2% (w/v) agarose gel. Adjust the shearing time and intensities, depending on the results of the DNA size analysis by agarose gel electrophoresis, to obtain an average size of 200-300 bp.
6. Determination of Free Versus Total DNA by Quantitative Real Time PCR (qRT-PCR)
- The ratio of nucleosome-free DNA (in the following referred to as free DNA) versus total DNA is determined by qRT-PCR17,18. Samples can also be quantitatively analyzed by microarray hybridization or deep sequencing for genome-wide determinations.
- Design qPCR primers for the regions to be tested and for positive and negative controls.
NOTE: Suitable positive controls are primers situated in the promoter of actively transcribed housekeeping genes such as ACTB, GAPDH, TPI1, or TUBA1A (encoding β-Actin, GAPDH, TPI or α-Tubulin). Appropriate negative controls are found in heterochromatin regions or gene deserts. Other suitable negative controls can be designed in genomic regions adjacent to the dynamically regulated locus under investigation to control for local copy number differences (Table 1). - Dilute the DNA samples to a final concentration of 10 ng/µL with TE buffer, and take the dilution factor into account for the final calculations (see step 6.5).
- Set up a qPCR reaction in triplicates in a 96-well plate, with the following reaction mix per well: template DNA: 20 ng (2 µL), forward primer 200 nM (0.4 µL of a 5 µM stock), reverse primer 200 nM (0.4 µL of a 5 µM stock), a commercial ready to use reaction mix containing green dye (5 µL from a 2x stock), and H2O to 10 µL.
- Run in a real time qPCR device, using the absolute quantification mode, and determine the Ct of the samples with the different primer pairs. Calculate the ratio of recovery of free DNA to total DNA for each primer using the following formula, taking into account the dilution factors from step 6.3:
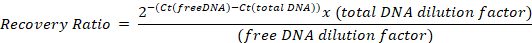
Representative results and an example calculation are given in Table 2. - To compensate for differences among samples, normalize to the positive control recovery ratio:
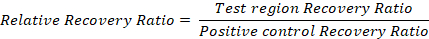
We have used the FAIRE method to measure differences in the chromatin accessibility of MHC class II genes in HEK293T cells as published19. MHC class II encoding genes are expressed in antigen presenting cells (APCs), either constitutively or in response to cytokine signaling20. The expression of MHCII genes is driven by an activator complex which binds to specific DNA elements, termed XY boxes, located in the promoter and enhancers of these genes21,22. This is reflected at the level of chromatin, as revealed by our FAIRE analysis of selected regions in the MHC class II genes HLA-DPB1 and HLA-DMA. These experiments showed that chromatin is open at the regions encompassing the XY boxes, while it is more compact in the gene bodies (Figure 2).
As an example, for the calculations, in this particular experiment, we diluted the total DNA sample 10 times (dilution factor 10), and the free DNA sample was not diluted (dilution factor 1) for the qRT-PCR. The Cts obtained for the different regions tested and the calculations to obtain the final recovery ratios and relative recovery ratios are listed in Table 2. These relative recovery ratios were obtained using the ACTB promoter as the positive control. Note that these calculations are for one of the replicates used to generate the results shown in Figure 2.
In a different context, chromatin accessibility was tested in a model for inducible gene expression. In this case, rapid changes of chromatin compaction in response to the pro-inflammatory stimulus tumor necrosis factor (TNF) were measured. HeLa cells were left untreated or stimulated with TNF for 1 h, followed by FAIRE to measure chromatin compaction at the promoter region controlling expression of the gene encoding the TNF-responsive cytokine interleukin 8 (IL8). These results showed a strongly increased chromatin accessibility at the IL8 promoter of TNF-stimulated cells (Figure 3). The accessibility at the IL-8 promoter after TNF stimulation is even higher than that found in the ACTB promoter showing a dramatic chromatin remodeling in this regulatory region. As the recovery ratio obtained in this case is higher than that obtained in the region used as the positive control (ACTB promoter) the relative recovery ratio is higher than one.
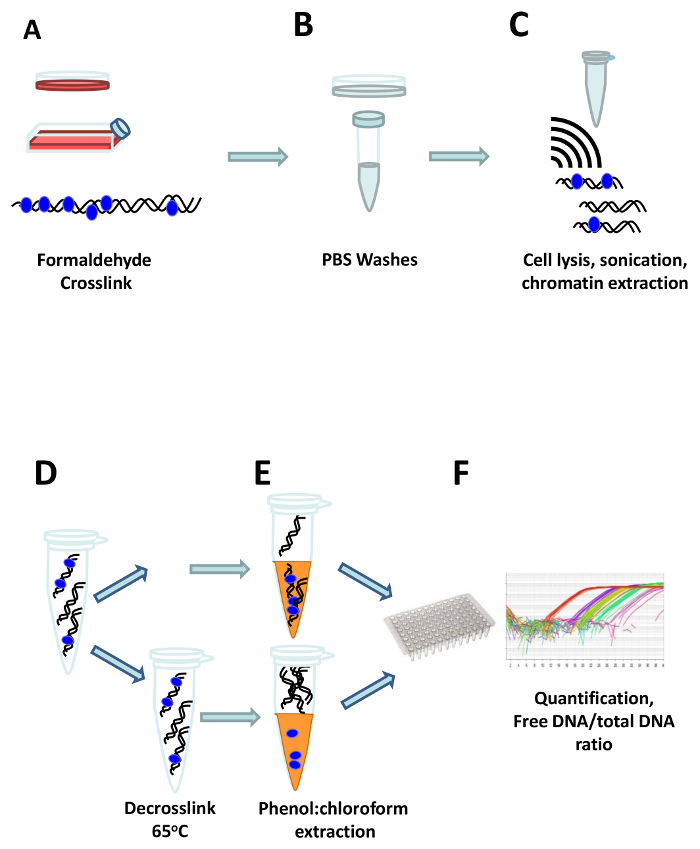
Figure 1: The FAIRE workflow. Cells are crosslinked directly in the cell culture flask or dish by adding formaldehyde (A). After quenching of the formaldehyde, cells are washed three times with ice cold PBS (B), and transferred to a reaction tube where they are resuspended in FAIRE lysis buffer and sonicated to shear the DNA (C). One aliquot of the extracted chromatin is de-crosslinked by heating at 65 °C, to get the total DNA reference (D), and afterwards the free DNA is extracted using phenol:chloroform both from the crosslinked and the de-crosslinked aliquots (E). Finally, the DNA is quantified, and the ratio of free vs. total DNA is calculated (F). Please click here to view a larger version of this figure.
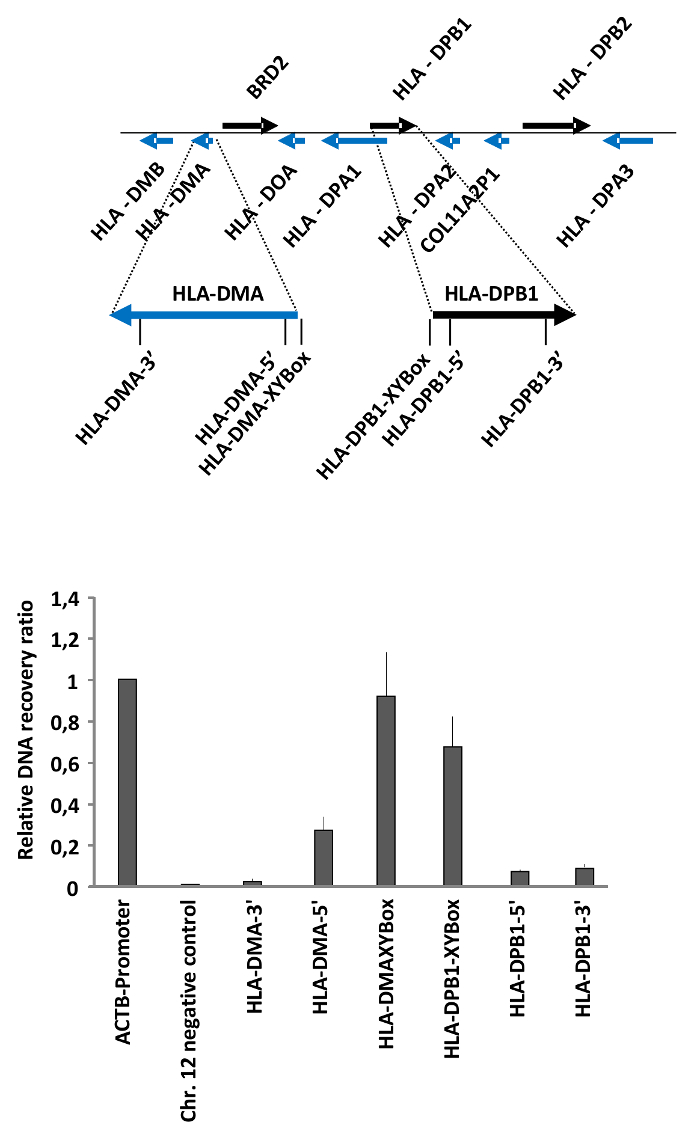
Figure 2: Determination of chromatin accessibility in the MHC class II gene cluster in HEK293T cells. HEK293T cells were analyzed by FAIRE. The ratio between free versus total DNA (i.e., the relative recovery ratio) was determined for the promoter of the ACTB gene as a positive control, a heterochromatin region on Chr. 12 as negative control, and the regulatory regions and gene bodies of the MHC class II genes HLA-DMA and HLA-DPB1. The upper part shows a schematic representation of the locus and the positions of the primers. Error bars represent standard deviations from two biological replicates measured in triplicates. The figure was modified from a published report19. Please click here to view a larger version of this figure.
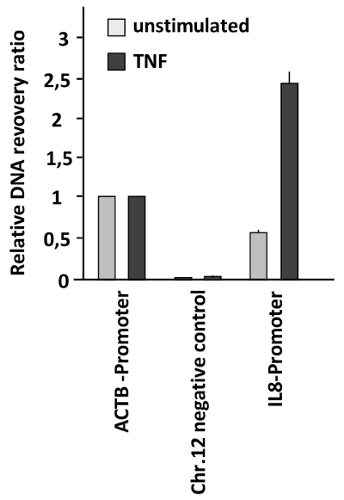
Figure 3: Changes of DNA accessibility at the IL8 promoter in response to TNF treatment. HeLa cells were stimulated with TNF (20 ng/mL) for 1 h and analyzed by FAIRE. The ratio between free versus total DNA was determined for the promoter of ACTB (positive control), a heterochromatin region on Chr. 12 (negative control), and the IL8 promoter. Error bars represent standard errors of the mean (SEM) from three biological replicates measured in duplicates. Please click here to view a larger version of this figure.

Figure 4: Optimization of the sonication conditions. Chromatin extracted from 293T cells was sonified using a focused sonicator with appropriate tubes (Table of Materials). The chromatin was sonicated for the indicated time with repetitions of 30 s with the following settings: peak power 150, duty factor 15, cycles per burst 500, followed by 30 s: peak power 2.5, duty factor 15, cycles per burst 500. The resulting chromatin was de-crosslinked, purified by phenol:chloroform extraction and loaded in a 2% agarose gel. An ethidium-bromide stained gel is shown, and the positions of the molecular weight markers are indicated in bp. In this experiment, the optimal chromatin size (200 – 300 bp) was obtained after 20 min of sonication. Please click here to view a larger version of this figure.
| Primer | Sequence |
| ACTB-Promoter-F | AAAGGCAACTTTCGGAACGG |
| ACTB-Promoter-R | TTCCTCAATCTCGCTCTCGC |
| Chr. 12 negative control-F | ATGGTTGCCACTGGGGATCT |
| Chr. 12 negative control- R | TGCCAAAGCCTAGGGGAAGA |
| HLA-DMA-XY-Box-F | CATCAGTCACTGGGGAGACG |
| HLA-DMA-XY-Box-R | GCTTCCCAGCCCAGTTACAT |
| HLA-DMA-5’-F | GGAGAGAACAATCTCCGCTTCA |
| HLA-DMA-5’-R | AGCTGCTATGTGTGGTTGGT |
| HLA-DMA-3’-F | TGGGGACCTAGTTAGGGAGC |
| HLA-DMA-3’-R | AGATCCATGGGAGGAGGCTT |
| HLA-DPB1-XY-Box-F | GTCCAATCCCAGGGTCACAG |
| HLA-DPB1-XY-Box-R | TGAAAAGAGCTGCAGTCAGGA |
| HLA-DPB1-5’-F | GCGTGTTCATGTCTGCATCC |
| HLA-DPB1-5’-R | TGATCCTCAGAGCCTGGACA |
| HLA-DPB1-3’-F | CCAGCCTAGGGTGAATGTTT |
| HLA-DPB1-3’-R | GCCTGGGTAGAAATCCGTCA |
| IL8 Promoter-F | GTGATGACTCAGGTTTGCCCT |
| IL8 Promoter-R | CTTATGGAGTGCTCCGGTGG |
Table 1: Primers for qPCR.

Table 2: Example calculations of FAIRE Relative Recovery Ratios. Please click here to download this file.
